 |
Главная Случайная страница Контакты | Мы поможем в написании вашей работы! | |
ВВЕДЕНИЕ 22 страница
|
|
5. Консолідація тромбу — його ущільнення, у результаті чого формується остаточний тромбоцитарний тромб. Відбувається в результаті в'язкого метаморфозу тромбоцитів та їх ретракції під впливом тромбостеніну.
26.3.5. Як відбувається адгезія тромбоцитів?
Основною причиною адгезії тромбоцитів є оголення (демаскування) колагену внаслідок ушкодження ендотелію судин.
Розрізняють доконтактну і контактну фази адгезії тромбоцитів.
Під час доконтактної фази в крові ще до контакту з ушкодженою стінкою судини відбувається первинна активація тромбоцитів. Спочатку змінюється форма тромбоцитів від дископодібної до сферичної (набряк). Потім викидаються довгі ниткоподібні відростки (від 3 до 10 у кожному тромбоциті).
Під час контактної фази відбувається взаємодія відростків активованих тромбоцитів з елементами базальної мембрани судинної стінки.
При цьому мають значення:
а) безпосередній контакт відростків тромбоцитів з колагеном;
б) опосередкований контакт тромбоцитів з колагеном через фактор Віллебранда;
в) реверсія електричного заряду інтими при її ушкодженні (заряд міняється з "—" на "+"), у результаті чого можлива електростатична взаємодія тромбоцитів, що мають негативний поверхневий заряд, зі стінкою судини;
г) уповільнення течії крові в ушкодженій судині.
26.3.6. Які фактори викликають агрегацію тромбоцитів? Яка динаміка цього процесу?
Причиною агрегації тромбоцитів є поява речовин-агрегантів. Вони можуть бути тромбоцитарного (виходять з активованих тромбоцитів) і нетромбоцитарного (вивільняються ушкодженими клітинами, лейкоцитами, утворюються в плазмі крові) походження.
Найбільше значення мають такі агреганти: а) АДФ. Вивільняється з ушкоджених клітин судинної стінки, гемолізованих еритроцитів, тромбоцитів у процесі їх активації;
б) тромбоксан А2 і арахідонова кислота. Тромбоксан А2 є продуктом циклоксигеназ-ного шляху перетворення арахідонової кислоти в тромбоцитах;
в) біогенні аміни — адреналін, серотонін. їхніми джерелами є плазма крові і тромбоцити;
г) фактор агрегації тромбоцитів (ФАТ). Є речовиною ліпідної природи. Вивільняється тканинними базофілами, базофілами і нейтрофілами крові;
ґ) тромбін. Є потужним агрегантом у дозах, значно менших, якщо порівнювати з тими, що викликають зсідання крові. Такі малі кількості тромбіну завжди утворюються в місцях ушкодження судинної стінки завдяки зовнішньому механізму зсідання крові, пов'язаному з вивільненням клітинного тромбопластину;
д) тромбоспондин. Вивільняється з активованих тромбоцитів, адсорбується на їхній мембрані, взаємодіє з білками крові.
Крім речовин-агрегантів, існують фактори, що мають антиагрегантні властивості. До них, зокрема, належать простациклін, продукти фібринолізу, простацикліноза-лежний макромолекулярний білок, білковий фактор Барнес-Ліана.
У процесі розвитку агрегації тромбоцитів розрізняють такі етапи:
1) початкова агрегація. Відбувається одночасно з адгезією тромбоцитів. її викликає АДФ нетромбоцитарного походження (вивільняється з ушкоджених клітин судинної стінки);
2) оборотна агрегація. На цьому етапі агрегація може бути припинена, тромбоцити ще не ушкоджені. Оборотну агрегацію обумовлюють тромбоцитарний АДФ, а також тромбоксан А, і арахідонова кислота;
3) необоротна агрегація. У цей період тромбоцити ушкоджуються і гинуть. Вважають, що основною причиною необоротної агрегації є тромбін, що утворюється локально.
26.3.7. Які механізми лежать в основі агрегації тромбоцитів?
І. Активація тромбоцитів (рис. 112). Речовини-агреганти збільшують проникність тромбоцитарної мембрани до іонів кальцію, у результаті чого концентрація останніх у цитоплазмі кров'яних пластинок зростає. Це викликає щонайменше чотири функціонально важливих ефекти:

Рис. 112. Механізм активації тромбоцитів
1) скорочення мікрофібрил, у результаті чого утворюються довгі ниткоподібні відростки;
2) посилення гідролізу АТФ, наслідком чого є утворення потужного агреган-та - АДФ;
3) викид гранул тромбоцитів;
4) активація фосфоліпази А2, що викликає утворення арахідонової кислоти, а потім і тромбоксану А.
II. Власне склеювання тромбоцитів. У цьому процесі мають значення:
а) утворення між тромбоцитами "містків", що складаються з АДФ та іонів кальцію;
б) білкове склеювання — утворення "містків", що складаються з білків плазми крові. Ці білки отримали назву плазмових кофакторів агрегації. До них відносять фібриноген, альбуміни, агрексони А і В. Усі ці білки склеюють тромбоцити завдяки взаємодії з глікопротеїновими рецепторами тромбоцитів (існує 5 типів таких рецепторів) і тромбоспондином — агрегантом, адсорбованим на тромбоцитарній мембрані.
26.3.8. Що являє собою процес зсідання крові? Які фактори беруть у ньому участь?
Зсідання крові (коагуляція) є складним багатоетапним ферментативним процесом, що в кінцевому підсумку закінчується утворенням фібринового згустку.
Основу процесу зсідання крові становлять реакції протеолізу, у яких прямо або опосередковано беруть участь 12 факторів зсідання (всі вони, за винятком ф. III, мають плазмове походження) і фактор 3 тромбоцитів.
До факторів зсідання крові відносять: ф. І— фібриноген, ф. П— протромбін* ф. Ш— тканинний тромбопластин; ф. IV— іони кальцію; ф. V— проакцелерин, ф. VII — проконвертин, ф. VIII — антигемофільний глобулін, ф. IX - фактор Кріст-маса, ф. X —фактор Стюарта—Прауера, ф. XI — плазмовий попередник тромбоплас-тину, ф. ХП - фактор Хагемана, ф. ХШ - фібринстабілізуючий фактор.
За функціональними властивостями всі фактори, що беруть участь у зсіданні крові, можна поділити на такі групи:
I. Білки-ферменти. Це в основному протеолітичні ферменти: ф. II, III, VII, IX, X, XI, XII. Один фактор (ф. ХШ) є трансферазою.
Усі зазначені ферменти містяться в крові і тканинах у неактивній формі. їх активація здійснюється шляхом протеолітичного відщеплення пептидів, що закривають активні центри ферментів. Таке відщеплення відбувається за участю активованого попереднього фактора зсідання (активної протеази). Таким чином, реакції активації зсідання крові мають каскадний, ланцюговий характер.
II. Неферментні білки- акцелератори (білки-прискорювачі). Такими є ф-V і ф.УШ. Ці фактори в сотні разів прискорюють ферментативні реакції зсідання крові. На відміну від ферментів вони споживаються в процесі коагуляції крові.
III. Фосфоліпідна матриця (мікромембрана), на якій відбуваються впорядковані реакції зсідання крові. Роль такої матриці можуть виконувати:
а) фактор 3 тромбоцитів;
б) фосфоліпідні фрагменти мембран ушкоджених клітин;
в) фосфоліпідні фрагменти мембран гемолізованих еритроцитів.
IV. Іони кальцію. їхня участь у зсіданні крові полягає у фіксуванні білкових факторів до фосфоліпідних матриць.
V. Субстрат зсідання крові - фібриноген (ф.І).
Більшість факторів зсідання синтезується в печінці. їх поділяють на дві групи:
а) вітамін-К-залежні: ф. П, VII, IX, X. Вітамін К у коферментній формі входить до складу печінкових карбоксилаз, які беруть участь в утворенні зазначених факторів;
б) вітамін-К-незалежнї. ф. І, V, XI. їх утворення не вимагає вітаміну К.
26.3.9. З яких фаз складається процес зсідання крові?
Процес зсідання крові проходить три фази.
І фаза - утворення протромбінази. Існує три механізми активації цього процесу (рис. 113):

Рис. 113. Механізми активації системи зсідання крові (ФЛ- фосфоліпіди)
а) зовнішній (тканинний) механізм. Активується при ушкодженні клітин. При цьому вивільняються ферменти - тканинний тромбопластин (ф. Ш) і фосфоліпідні фрагменти мембран, що стають матрицею, на якій фіксуються фактори зсідання крові. Це дуже швидкий механізм активації зсідання. Він забезпечує зсідання крові поза кровоносними судинами (при крововиливах у тканини) і локальне утворення малих доз тромбіну, необхідного для необоротної агрегації тромбоцитів (рис. 114);
б) внутрішній (кров 'яний) механізм. Початком його є контактна або ферментативна активація ф. XII крові. Внутрішній механізм зсідання досить тривалий, тому перша його фаза по суті визначає час усього процесу коагуляції крові;
в) макрофагально-моноцитарний механізм. На відміну від двох попередніх є механізмом патологічної активації зсідання крові. Його викликає дія на макрофаги ендотоксинів бактерій, імунних комплексів, комплементу, продуктів розпаду тканин. При цьому з макрофагів вивільняється уже активна протромбіназа (ф.Ха).

Рис. 114. Зовнішній і внутрішній механізми активації зсідання крові
Цей механізм має певне пристосувальне значення, оскільки завдяки зсіданню крові обмежується поширення патогенних факторів в організмі. її фаза — утворення тромбіну. Відбувається за участю активної протромбінази іф.У(рис.П5).

Рис. 115.7/ фаза зсідання крові
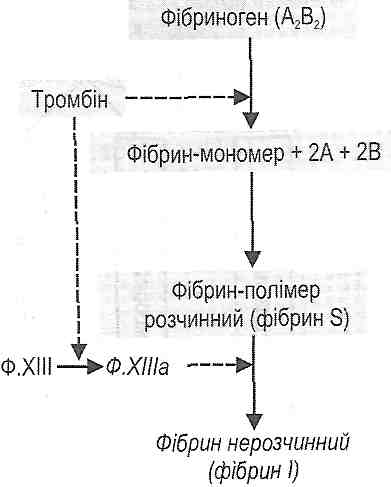
III фаза - утворення фібрину. Складається з кількох послідовних етапів (рисі 16):
а) утворення фібрину—мономеру із фібриногену під дією тромбіну;
б) утворення розчинного фібрину-полімеру (фібрину S);
в) утворення нерозчинного фібрину (фібрину І) під дією активного ф. XIII.
26.3.10.Як класифікують порушення гемостазу?
Порушення гемостазу (гемостазіопатії) поділяють на три групи (рис. 117):

Рис. 117. Класифікація порушень гемостазу
I. Геморагічні гемостазіопатії — геморагічні діатези.
II. Тромбогеморагічні гемостазіопатії- синдром дисемінованого внутрішньосудин-ного зсідання крові (ДВЗ-синдром),
III. Тромбофілічні гемостазіопатії — тромбози і тромбоемболії.
26.3.11. Що таке геморагічні діатези? Як їх класифікують?
Геморагічний діатез — це схильність організму до повторних кровотеч і крововиливів, що виникають спонтанно або після незначних травм. Геморагічні діатези поділяють на дві великі групи (рис. 118).

Рис. 118. Класифікація геморагічних діатезів
I. Порушення судинно-тромбоцитарного гемостазу:
а) вазопатії)
б) тромбоцитопенії;
в) тромбоцитопатії.
II. Порушення коагуляційного гемостазу - коагулопатії.
26.3.12. Що таке вазопатії? Як їх класифікують?
Вазопатії—ц& спадково обумовлені або набуті геморагічні діатези, що виникають як наслідок первинних порушень судинної стінки.
Залежно від механізму розвитку вазопатії поділяють на дві групи:
1) запальні вазопатії— васкуліти;
2) диспластичні вазопатії— ураження судин, пов'язані з порушеннями їхньої сполучної тканини (неповноцінність судинної стінки).
26.3.13. Які етіологія і патогенез запальних вазопатій?
Залежно від причин виникнення запальні вазопатії поділяють на:
1) інфекційні васкуліти. Є проявом цілого ряду інфекційних захворювань (вірусних геморагічних гарячок, висипного тифу, сепсису);
2) імунні васкуліти. Розвиваються як наслідок імунокомплексних захворювань (алергічних реакцій III типу за класифікацією Кумбса і Джелла), наприклад, при системному червоному вовчаку, вузликовому періартеріїті, геморагічному васкуліті (хвороба Шенляйн-Геноха);
3) інфекційно-імунні васкуліти. Поєднують обидва попередніх механізми.
У патогенезі запальних вазопатій провідна роль належить ушкодженню ендотелію, що може бути обумовлено:
а) цитопатичною дією ендотеліотропних вірусів;
б) токсинами мікробів, наприклад веротоксином, що його виділяють бактерії кишкової групи; *
в) комплексами антиген-антитіло і комплементом.
Наслідком ушкодження ендотелію судин є:
1) діапедез еритроцитів, що клінічно виявляється точковими крововиливами (пете-хіями);
2) інтенсивне мікротромбоутворення, що викликає порушення мікроциркуляції і живлення тканин;
3) тромбоцитопенія споживання (результат утворення мікротромбів).
26.3.14. Які етіологія і патогенез диспластичних вазопатій?
В основі диспластичних вазопатій лежать набуті або спадково обумовлені порушення сполучної тканини стінки кровоносних судин. Прикладами таких порушень є:
1. Гіповітаміноз С. Аскорбінова кислота— необхідний компонент реакції гідро-ксилювання проліну, унаслідок якої він перетворюється в оксипролін. Ця реакція вважається однією з ключових в утворенні колагену. При гіповітамінозі С з урахуванням сказаного порушується утворення повноцінного колагену (ламкість судин, випадання зубів і т. д.).
2. Телеангіектазії. Це спадково обумовлені локальні дефекти сполучної тканини судин, що обумовлюють стоншення їх стінок і розширення просвіту. Телеангіектазії є джерелом небезпечних для життя кровотеч, особливо при локалізації у внутрішніх органах.
3. Гемангіоми. Це судинні пухлини, які часто кровоточать.
4. Синдром Елерса-Данло. Його основу складають генетично обумовлені дефекти ] колагену.
У патогенезі диспластичних вазопатій мають значення:
1) стоншення стінок мікросудин і розширення їхнього просвіту;
2) неповноцінний локальний гемостаз. Через недостатню кількість або неповноцінність колагену в субендотелії судин порушується адгезія тромбоцитів;
3) легка ранимість судин.
26.3.15. Що таке тромбоцитопенія? Які механізми можуть лежати в основі розвитку тромбоцитопенії?
Тромбоцитопенія — це зменшення вмісту тромбоцитів в одиниці об'єму периферичної крові нижче 150 109/л.
На думку багатьох авторів, геморагічні прояви тромбоцитопенії з'являються при зменшенні кількості тромбоцитів нижче 50 109/л.
За походженням тромбоцитопенії можуть бути спадково обумовленими і набутими.
За механізмом розвитку виділяють такі види тромбоцитопенії. І. Тромбоцитопенії, пов'язані з порушеннями утворення тромбоцитів:
а) мієлотоксичні тромбоцитопенії— виникають унаслідок ушкодження кровотворних клітин. Дуже часто поєднуються з анемією й лейкопенією. Причинами їх розвитку є ті самі фактори, які викликають розвиток гіпопластич-ної анемії;
б) дефіцитні тромбоцитопенії— обумовлені недостатністю вітаміну В12 або фолієвої кислоти;
в) дисрегуляторні тромбоцитопенії— пов'язані з порушенням утворення тромбоцитопоетинів — речовин, що стимулюють утворення тромбоцитів;
г) тромбоцитопенії, пов'язані зі зменшенням плацдарму кровотворення, — розвиваються при лейкозах і метастазах злоякісних пухлин.
її. Тромбоцитопенії, пов'язані з посиленим руйнуванням тромбоцитів. Причиною такого руйнування можуть бути:
а) імунне ушкодження, обумовлене антитромбоцитарними антитілами на власні компоненти кров'яних пластинок або на лікарські препарати, адсорбовані на тромбоцитах. Аутоімунне ушкодження вважають найбільш імовірним механізмом розвитку так званої ідіопатичноїтромбоцитопенічноїпурпури (хвороби Верльгофа);
б) ггперспленізм — гіперфункція селезінки, що супроводжується часто спле-номегалією. У результаті підвищення фагоцитарної активності макрофагів відбувається інтенсивне руйнування всіх формених елементів крові, у тому числі й тромбоцитів;
в) механічне ушкодження тромбоцитів. Часто виникає при гемангіомах і накладенні штучних клапанів серця;
г) набутімембранопатії (гемолітична анемія Маркіафави—Мікеллі). Соматичні мутації кровотворних клітин спричиняються до утворення пулів клітин
(еритроцитів, гранулоцитів, тромбоцитів) з дефектами мембрани. У результаті збільшується чутливість таких клітин до дії комплементу й відбувається їх руйнування. III. Тромбоцитопенії споживання. Виникають у результаті посиленого використання тромбоцитів на утворення тромбів (хвороба Шенляйн—Геноха, хвороба Мошковича, ДВЗ-синдром).
26.3.16. Який патогенез порушень гемостазу в умовах тромбоцитопенії?
У патогенезі геморагічного синдрому при тромбоцитопеніях мають значення:
1) порушення ангіотрофічної функції тромбоцитів, у результаті чого виникають дистрофічні зміни в ендотелії і збільшується ламкість мікросудин. Це веде до збільшення ранимості судин, діапедезу еритроцитів, крововиливів. Останні виявляють себе петехіями на шкірі, кровотечами з ясен і носа, крововиливами в головний мозок і сітківку ока;
2) порушення адгезії й агрегації тромбоцитів. Це викликає порушення формування тромбоцитарного тромбу й призводить до збільшення часу кровотечі (проба Дюка);
3) порушення вторинного спазму ушкоджених артеріол. При тромбоцитопеніях вивільняється мало біогенних амінів (катехоламінів, серотоніну), з дією яких пов'язане скорочення гладких м'язів судин;
4) порушення зсідання крові. Обумовлено недостатнім вивільненням фактора 3 пластинок і тромбостеніну. У результаті порушується І фаза зсідання крові і ретракція згустку.
26.3.17. Що таке тромбоцитопатії? Як їх класифікують?
Тромбоцитопатії— це порушення функціональних властивостей тромбоцитів, йґ якісна неповноцінність. При цьому кількість тромбоцитів може залишатися в нормі J
За походженням тромбоцитопатії бувають спадково обумовленими і набутими.
За характером якісних дефектів кров'яних пластинок тромбоцитопатії поділяють на ендо- і екзотромбоцитарні.
Ендотромбоцитарні тромбоцитопатії обумовлені порушеннями складових частин тромбоцитів. їх, у свою чергу, поділяють на мембранопатії, гранулопатії і фер-ментопатії. Мембранопатії виникають при спадкових аномаліях мембранних пгіко-протеїнів, що виконують функції клітинних рецепторів; при блокаді цих рецепторів аномальними білками плазми крові (парапротеїнами), при ушкодженні мембрани кров'яних пластинок патогенними факторами. Гранулопатії виявляються дефіцитом гранул І і II типів. В основі ферментопатії може лежати зменшення активності ферментів циклу Кребса, гліколізу, порушення функцій АТФ-аз, циклоксигенази і тром-боксансинтетази.
При екзотромбоцитарних тромбоцитопатіях причини порушення функцій тромбоцитів лежать поза кров'яними пластинками. У зв'язку з цим екзотромбоцитарні тромбоцитопатії можуть бути:
а) пов'язаними зі змінами плазми крові (дефіцит плазмових білків, що є плазмовими кофакторами агрегації тромбоцитів);
б) пов'язаними зі змінами в судинній стінці (порушення утворення фактора Вілле-бранда ендотелієм судин, розлади зовнішнього механізму зсідання крові). Залежно від сутності порушень гемостазу виділяють:
1) тромбоцитопатії з первинним порушенням адгезії тромбоцитів^
2) тромбоцитопатії з первинними порушеннями агрегації тромбоцитів;
3) тромбоцитопатії з первинним порушенням реакцій вивільнення вмісту тромбоцитів',
4) тромбоцитопатії, пов'язані з дефіцитом або зменшенням доступності фактора З тромбоцитів.
26.3.18. Наведіть приклади тромбоцитопатій з різними механізмами порушень гемостазу.
1. Тромбоцитопатії з первинним порушенням адгезії тромбоцитів:
а) хвороба Віллебранда — ангіогемофілія. Обумовлена генетичними порушеннями синтезу фактора Віллебранда ендотеліальними клітинами (тип спадкування аутосомно-домінантний);
б) хвороба Бернара—Сульє — макротромбоцитодистрофія. Причиною є спадково обумовлений дефект глікопротеїнів тромбоцитарної мембрани (ГШЬ), що взаємодіють з фактором Віллебранда. При цьому тромбоцити набувають гігантських розмірів. Тип спадкування аутосомно-рецесивний.
2. Тромбоцитопатії з первинними порушеннями агрегації тромбоцитів - диза-грегаційні. Найчастіше буває тромбастенія Гланцмана, що виникає як наслідок дефектів мембранних глікопротеїнів І і II типів, що беруть участь в агрегації. При цьому адгезія тромбоцитів і вивільнення їхніх гранул відбуваються, а агрегація -ні, незважаючи на дію таких потужних агрегантів, як АДФ, адреналін, тромбін. Тип спадкування аутосомно-рецесивний.
3. Тромбоцитопатії з первинним порушенням реакцій вивільнення вмісту тромбоцитів:
а) порушення дегрануляції тромбоцитів — "парез реакції вивільнення ". Виникає, зокрема, при порушенні утворення тромбоксану А2 при дії ацетилсаліцилової кислоти. Показано, одо одноразове приймання аспірину необоротно збільшує час вивільнення гранул від 3,5 до 6 діб, поки не з'являться нові тромбоцити;
б) недостатність накопичення й збереження вмісту гранул тромбоцитів. Виникає, як правило, при генетично обумовлених порушеннях.
Розлади реакцій вивільнення гранул викликають порушення другої хвилі агрегації тромбоцитів. Початкова агрегація кров'яних пластинок закінчується їх дезагрегацією.
4. Тромбоцитопатії, пов'язані з дефіцитом або зменшенням доступності фактора 3 пластинок. У їхній основі можуть лежати або генетично обумовлені дефекти структури цього фактора, або порушення його вивільнення з ушкоджених тромбоцитів. При цьому порушується зсідання крові (коагуляційний гемостаз). Адгезив-но-агрегаційні властивості тромбоцитів не міняються.
26.3.19. Чим можуть бути обумовлені порушення коагуляційного гемостазу - коагулопатії?
В основі розвитку коагулопатій можуть бути:
1) зменшення активності системи зсідання крові;
2) підвищенням активності антикоагулянтної системи;
3) збільшенням активності фібринолітичної системи.
26.3.20. Що може бути причиною безпосереднього порушення І фази зсідання крові?
Залежно від характеру порушень І фази зсідання крові виділяють три групи розладів.
I. Ізольовані порушення зовнішнього механізму активації зсідання. Виникають при дефіциті ф. VII - гіпопроконвертинемії. Цей дефіцит може бути спадково обумовленим (тип спадкування аутосомно-рецесивний) або набутим (гіповітаміноз К, ураження печінки).
II. Ізольовані порушення внутрішнього механізму активації зсідання:
а) дефіцит ф.УШ- гемофілія А. Найчастіше виникає як генетичний дефект коагулянтної частини ф.УШ (тип спадкування зчеплений з Х-хромосомою). Можливе утворення аутоантитіл проти білкових компонентів цього фактора зсідання;
б) дефіцит ф. IX- гемофілія В. Причиною розвитку є спадкова патологія (тип спадкування зчеплений з Х-хромосомою), дефіцит вітаміну К або ураження печінки, антитіла проти ф. IX;
в) дефіцит ф. XI— гемофілія С. Виникає при генетичних порушеннях (тип спадкування аутосомно-рецесивний) або ураженнях печінки;
г) дефіцит ф. ХП. Спадкова патологія, що буває дуже рідко. Завдяки калікре-їн-кініновій системі цей дефект добре компенсується, оскільки запуск внутрішнього механізму зсідання відбувається через зовнішній;
ґ) дефіцит ф. З тромбоцитів. Є наслідком тромбоцитопенії або певних видів тромбоцитопатій.
III. Поєднані порушення зовнішнього і внутрішнього механізмів зсідання. Розвиваються при дефіциті ф. X, що може бути спадково обумовленим (тип спадкування аутосомно-рецесивний) або набутим (гіповітаміноз К, ураження печінки).
26.3.21. Що може бути причиною безпосереднього порушення II фази зсідання крові?
1. Дефіцит ф.ІІ— гіпопротромбінемія. Найчастіше має набутий характер і розвивається внаслідок гіповітамінозу К або уражень печінки.
2. Дефіцит ф. V— парагемофілія. Порушення утворення проакцелерину може бути обумовлено ураженнями печінки або аутоантитілами проти ф.У
26.3.22. Що може бути причиною безпосереднього порушення III фази зсідання крові?
1. Дефіцит фібриногену:
а) афібриногенемія - повна відсутність фібриногену (спадкове захворювання з аутосомно-рецесивним типом спадкування);
б) гіпофібрииогенемія - зменшення синтезу фібриногену в печінці при її ураженнях.
2. Дисфібршшгенемії - якісні порушення фібриногену. Розвиваються як наслідок генетичних дефектів (тип спадкування аутосомно-домінантний). Виявляються утворенням аномального фібрину.
3. Порушення полімеризації фібрину. Розвиваються б результаті утворення комплексів фібриногену з фібрином-мономером і проміжними продуктами, від яких ще повністю не відщепились пептиди А і В. При цьому утворюється так званий "заблокований фібриноген" ("тромбінрезистентний фібриноген"), що не піддається дії тромбіну.
4. Дефіцит ф. ХШ. Виникає як наслідок спадкових порушень (тип спадкування ау-тосомно-рецесивний). виявляється порушеннями перетворення розчинного фібрину (фібрину S) у нерозчинний (фібрин І).
26.3.23. Які речовини складають антикоатулянтну систему крові?
І. Первинні антикоагулянти. Постійно синтезуються в організмі й тому завжди містяться в плазмі крові. До них відносять:
1) антитромбін ПІ- основний універсальний антикоагулянт, що є інгібітором протеаз. Синтезується ендотелієм судин. Пригнічує активність усіх протеолітичних ферментів крові, у тому числі тромбіну, калікреїну, плазміну, ф. ХІІа, ф. ХІа, ф. Ха, ф. ІХа, ф.УІІа;
2) гепарин (антитромбін II) — глікозаміноглікан, що вивільняється тканинними базофілами і базофілами крові при їх дегрануляції. Антикоагулянтні властивості має не сам гепарин, а комплекс гепарину з антитромбіном III. Завдяки гепарину антитромбін III фіксується на поверхні ендотелію судин, де його антикоагулянтні властивості в багато разів зростають;
3) а -антитрипсин, а2-макроглобулін, інгібітор СІ-компонента комплементу - усі вони є неспецифічними інгібіторами протеаз, у тому числі й факторів зсідання крові.
її. Вторинні антикоагулянти. У плазмі крові в нормі не містяться. Утворюються в процесі зсідання крові й фібринолізу. До них відносять:
1) антитромбін І— фібрин, що адсорбує і в такий спосіб інактивує велику кількість тромбіну;
2) продукти фібринолізу. Перешкоджають процесам полімеризації фібрину і утворенню фібринових структур.
26.3.24. Чим може бути обумовлене підвищення активності антикоагулянтної системи крові?
Підвищення активності антикоагулянтної системи крові закономірно виникає при:
1) збільшенні вмісту гепарину в крові — гіпергепаринеміг. Це може бути обумовлено посиленою дегрануляцією тканинних базофілів і базофілів крові, зокрема, при алергічних реакціях І типу за класифікацією Кумбса і Джелла, руйнуванням базо-фільних лейкоцитів при лейкозах, введенням гепарину ззовні;
2) появою "патологічних " антикоагулянтів, до яких відносять антитромбін V, що порушує полімеризацію фібрину-мономеру; "вовчаковий" антикоагулянт, що порушує утворення протромбіназного комплексу; парапротеїни, що унеможливлюють полімеризацію фібрину.
26.3.25. Що входить у поняття "фібринолітична система"?
Фібриноліпшчна система — це система, яка забезпечує лізис (протеоліз) фібрину в кровоносному руслі. У такий спосіб вона бере участь у підтримці рідкого стану крові й у відновленні кровообігу в тромбованих судинах.
До складу системи фібринолізу входять (рис. 119):

Рис. 119. Схема фібринолізу
1) плазміноген (профібринолізин) — неактивний протеолітичний фермент, що завжди міститься в плазмі крові;
2) плазмін (фібринолізин) — активна форма плазміногену. Утворюється в результаті дії активних протеаз на плазміноген і відщеплення від його молекули пептиду, який "закриває" активний центр;
3) активатори фібринолізу — велика група речовин, які або самі є протеазами і здатні перетворювати плазміноген у плазмін, або викликають появу таких протеаз;
4) інгібітори фібринолізу. До них відносять інгібітори протеаз, серед яких найбільше значення має а2-антиплазмін.
Розрізняють внутрішній і зовнішній механізми активації фібринолізу.
Внутрішній механізм обумовлений активацією фактора XII зсідання крові й утворенням калікреїну, унаслідок чого в крові з'являється велика кількість активаторів фібринолізу.
Зовнішній механізм пов'язаний з надходженням у кров готових активаторів фібринолізу: ендотеліального, клітинного, тканинного (урокіназа), бактеріального (стрептокіназа).
Дата публикования: 2014-11-04; Прочитано: 1468 | Нарушение авторского права страницы | Мы поможем в написании вашей работы!
