 |
Главная Случайная страница Контакты | Мы поможем в написании вашей работы! | |
Миотонии
|
|
^TJ^a Z гетерогенная Wnna нервно-мышечных заболеваний, объеди-
енная общим характерным комплексом нарушений мышечного тонуса,
о являющимся затруднением расслабления мышц после активного сокра-
сличают наследственные миотонии (стационарные медленно про-Фующие и периодические, рецидивирующие формы) и миотоничес-
ми мнijiромы.
Врожденная миотония (болезнь Лейдена—Томсева). За! ие впе
рвые описано Лейденом в 1874 г. Томсен в 1876 г. обратил внимание на н следственную природу болезни на примере своей семьи (дети и м ственники — 20 членов его семьи в 4 поколениях страдали Mi-Частота 0,3—0,7 на 100 000 населения. Наследуется по аутосомно-^ нантному типу. Пенетрантность более высокая у лиц мужского пола.
Патогенез. Имеют значение нарушения проницаемости ной мембраны, изменение ионного и медиаторного обмена (наруш функциональной взаимосвязи в звене кальций—тропонин—актомиозин). повышенная чувствительность ткани к ацетилхолину и калию.
Патоморфология. При световой микроскопии обнаруживается гипертрофия отдельных мышечных волокон; гистохимия ее ки определяется уменьшение размеров II типа мышечных волокон; при электронной микроскопии выявляются умеренная гипертрофия саркоплазматической сети, изменение формы и увеличение размера митохондрий, расширение телофраг-мы миофибриллярных волокон.
Клинические проявления. Впервые симптомы заболевания проявляются преимущественно в возрасте 8—15 лет. Ведущими признаками служат миотонические спазмы — затруднения расслабления мыши после активного напряжения. Миотонические спазмы локализуются в различных группах мышц, чаще в мышцах кисти, ног, жевательных мышцах и круговых мышцах глаза. Сильное сжатие пальцев кисти, длительное статическое напряжение ног, смыкание челюстей, зажмуривание глаз вызывают тонические спазмы. Фаза расслабления мышц задерживается на продолжительное время, и больные не в состоянии быстро разжать кисти, изменить положение ног, открыть рот, глаза. Повторные движения уменьшают миотонические спазмы. Повышение механической возбудимости мышц определяется с помощью специальных приемов: при ударе неврологическим молоточком по возвышению I пальца происходит приведение его к кисти (от нескольких секунд до минуты) — «симптом большого пальца», при ударе перкуссионным молоточком по языку на нем появляется ямка, перетяжка — «симптом языка». Внешний вид больных своеобразен. Вследствие диффузных гипертрофии различных мышц они напоминают профессиональных атлетов. При пальпации мышцы плотные, твердые, однако объективно мышечная сила снижена. Сухожильные рефлексы нормальны, в тяжелых случаях снижены.
Течение. Болезнь медленно прогрессирует. Трудоспособность сохраняется в течение длительного времени.
Диагностика и дифференциальный диагноз. Диагноз строится на основании генеалогического анализа (аутосомно-доми-нантный тип наследования), особенностей клинической картины (атлеп ческий тип телосложения, диффузные гипертрофии мышц, миотоническт синдром), данных глобальной электромиографии (миотоническая реакция).
Дифференцировать заболевание следует от других форм миот< иногда — от псевдогипертрофических форм прогрессирующих мышечных дистрофий.
Лечение. Назначают дифенин (по 0,1 -0,2 г 3 раза в лень в течение 2-3 нед), диакарб (по 0,125 г 2 раза в день в течение 2—3 нед). гтреп; кальция (внутривенно 10 % раствор хлорида кальция по 10 мл или г нат кальция внутримышечно). Предполагается, что дифенин оказывает
 пплисинаптическое проведение в ЦНС,
пплисинаптическое проведение в ЦНС,
тормозящее влияние на моно- и по._ Целесообразны физиотерапия
а диакарб юменяетпронида^остъмемр. ем^ лечебная гимнас-
в виде гальванического воротника и трусив
тика. Россолимо—Штейнерта—Куршмана. Заболе-
Дистрофическая миотония rut ^ ^^ ^ ^ впоследствии Штейнер-
вание впервые описано iГ.И. ^^ ___- юО 000 населения. Наследует-
том и Кглгшгманом в 1912 г. частом
| кон, разрастание |
^ определяются из-
СаРТлаи"аи «ТкиТп роявления. Первые признаки заболевания проявляются в 10-20-летнем возрасте. Характерно сочетание миотоничес-Г»вш, нейроэндокринных, сердечно-сосудистых нарушении. ХоГнический симптомокомплекс, как и при врожденной миотонии Том-сена проявляется миотоническими спазмами, повышенной механической возбудимостью. Степень выраженности миотонического феномена в поздних стадиях болезни при выраженной дистрофии мышц ослабевает. Мио-патический синдром характеризуется патологической мышечной утомляемостью, слабостью, мышечными атрофиями, которые локализуются преимущественно в мышцах лица, шеи, дистальных отделов конечностей. Вследствие атрофии внешний вид больных своеобразен: голова опущена на шею, лицо амимичное, худое, особенно в височных областях, веки полуопущены, ноги"й~руки сужены в дистальных отделах. Типичны «выеденные» стопы, «обезьяньи» кисти. Походка перонеальная («степпаж»), иногда при атрофиях проксимальных групп мышц с компонентом «утиной». Мышечный тонус снижен, сухожильные рефлексы рано угасают. Нейроэндо-кринные расстройства многообразны. Наиболее выражены изменения в гонадах. У мужчин часто наблюдаются крипторхизм, снижение либидо, импотенция, у женщин — нарушения менструального цикла. У многих больных отмечаются раннее облысение, истончение и сухость кожи. Сердечнососудистые расстройства постоянны. Имеются полная или частичная блокада ножек пучка Гиса, низкий вольтаж на ЭКГ, аритмия. Заболевание медленно прогрессирует.
Диагностика и дифференциальный диагноз. Диагноз ставится на основании данных генеалогического анализа (аутосомно-доминантный тип наследования), особенностей клиники (сочетание мио-тонических, миопатических, нейроэндокринных, сердечно-сосудистых нарушений), результатов глобальной электромиографии (миотоническая реакция), биохимического исследования крови (инсулинорезистентность).
Дифференцировать заболевание следует от врожденной миотонии Томсена, других миотонических форм, прогрессирующих мышечных дистрофий — дистальной миопатии, невральной амиотрофии.
Лечение. Как и при врожденной миотонии, положительный эффект дают дифенин, диакарб. Показано применение анаболических стероидов (ретаболил, неробол, метиландростендиол). В диете следует уменьшить содержание калия.
24.2. Пирамидные и экстрапирамидные дегенерации 24.2А. Семейный спастический паралич Штрюмпеля
Хроническое прогрессирующее наследственно-дегенеративное заболевание нервной системы, характеризующееся двусторонним поражением пирамидных путей в боковых и передних канатиках спинного мозга. А. Штрюмпель в 1866 г. отметил семейный характер болезни. Применяется также название «семейная спастическая параплегия Эрба—Шарко—Штрюмпеля».
Этиология и патогенез. Заболевание является наследственным, чаще передается по аутосомно-доминантному, реже — по аутосомно-рецессивному и сцепленному с полом (с Х-хромосомой) типу. Патогенез дегенерации и первичный биохимический дефект неизвестны.
П атоморфология. Наиболее часто поражаются поясничная и грудная части спинного мозга, реже — ствол головного мозга. Отмечается симметричное глиозное перерождение пирамидных путей в боковых и передних канатиках, пучках Голля. Описаны случаи дегенеративных изменений в клетках коры передней центральной извилины, передних рогов спинного мозга, мозжечковых проводниках.
Клинические проявления. Развитие заболевания постепенное. Наиболее часто первые симптомы появляются во втором десятилетии жизни, хотя отмечаются большие колебания возраста, в котором начинается болезнь. Вначале возникают скованность в ногах и быстрая утомляемость при ходьбе, нарастающие по мере прогрессирования заболевания. Развивается характерная спастическая походка, присоединяются варусная и эквиноварусная деформации стоп, изменения стоп по типу «стопы Фрид-рейха», сухожильные и мышечные контрактуры, особенно в голеностопных суставах. Постепенно слабость в нижних конечностях нарастает, однако полного паралича нижних конечностей не наблюдается. При клиническом обследовании больных уже в начальных стадиях заболевания обнаруживается повышение сухожильных рефлексов, рано появляются патологические рефлексы сгибательной и разгибательной групп (Бабинского, Оппенгейма, Россолимо, Гордона, Шеффера, Бехтерева—Менделя, Жуковского), клонусы стоп, коленных чашечек. Кожные рефлексы в большинстве случаев сохраняются, функции тазовых органов не нарушены. Расстройства чувствительности отсутствуют. Интеллект сохранен. Значительно позже в патологический процесс вовлекаются верхние конечности. Нередко к нижнему спастическому парапарезу присоединяются симптомы поражения зрительных и глазодвигательных нервов, нистагм, дизартрия, атаксия и интенци-онное дрожание.
Диагностика и дифференциальный диагноз. Диагноз обычно не вызывает затруднений при наличии семейного характера заболевания и типичной клинической картины.
В атипичных спорадических случаях заболевание следует отграничивать от спинальной формы рассеянного склероза, бокового амиотрофичес кого склероза, опухолей спинного мозга и других патологических процессов различной этиологии вызывающих компрессию спинного мозга, а также фуникулярного миелоза, нейросифилиса и других форм мозжечке пирамидных дегенерации Для спинальной формы рассеянного склероз, "аряду с нижним спастическим парапарезом характерны ремиттирующее течение, непостоянство и временная обратимость отдельных симптомов,
v „„гянов выпадение или асимметрия брюш-
„арушение функций тазо^'хя°сРиГмп°Томов поражения в целом, изменение
„ых рефлексов и асимметрия симптом°«Р нальной ЖИДКости. Ре-
иммунологических "°>™^^ледственном характере заболевания,
шающее значение имеют Данные о^нас д а болезнь Штрюмпеля
В отличие от бокового ами0?°0Фт^ству^изнаки поражения перифе-
начинается в ^"^"^спшу^ш подергивания, атрофия мелких
рического мотонеирона (фасцикулярн б х расстройств. При
мышц кисти, характерные «зме^"и«f1 ог/' лей и синдрома компрессии
дифференциации от Э5с^а™и ИМ™ т значение сегментарные расстрой-
спинного мозга другой ™r™"жения конечностей, наличие блока
ства чувствительности ^имме^ия " ffl диссоциация в це-
SS^SS"прТлюмбальной пункции, характерные для При «йросифилисе в отличие от болезни Штрюмпеля в анамне-тся указания на кожные проявления. Ведущими в клинической картине являются симптомы поражения задних канатиков спинного мозга, определяются характерные зрачковые расстройства, изменения в крови, цереброспинальной жидкости.
Дифференциальная диагностика семейной спастической параплегии с другими дегенеративными поражениями спинного мозга бывает иногда затруднительной. Помогает выявление симптомов поражения других отделов нервной системы (мозжечковых, глазных и др.).
Течение и прогноз. Течение заболевания медленно прогрессирующее; отмечается более злокачественное течение при возникновении его в раннем возрасте. При позднем развитии болезни гипертония и гиперрефлексия преобладают над двигательными нарушениями. Прогноз для жизни благоприятный. Степень утраты трудоспособности зависит от выраженности нарушения функций нервной системы.
Лечение. Симптоматическое. Назначают препараты, снижающие мышечный тонус, — мидокалм, баклофен, изопротан (скутамил), транквилизаторы: сибазон (седуксен), нозепам (тазепам), хлозепид (элениум). Показаны физиотерапевтические процедуры, парафиновые аппликации на мышцы нижних конечностей. Применяются точечный массаж, рефлексотерапия, лечебная физкультура, при необходимости — ортопедические мероприятия. Показаны курсы общеукрепляющего лечения: витамины группы В, метаболические препараты: пирацетам (ноотропил), пиридитол (эн-цефабол), аминалон, церебролизин, аминокислоты, АТФ, кокарбоксилаза, препараты, улучшающие микроциркуляцию.
24.2.2. Болезнь Паркинсона
[аболевание впервые описано английским врачом Джеймсом Паркинсо-ном, который назвал его дрожательным параличом. В 1877 г. Жан Мартен Шарко дополнил клиническую характеристику болезни Заболевание встречается у 60-140 на 100 000 населения; частота его резко увеличивается с возрастом. Согласно статистическим данным, дрожательный паралич встречается у 1 % населения до 60 лет и у 5 % более старшего возраста. Мужчины болеют несколько чаще, чем женщины.
Этиология и патогенез. Клинические проявления дрожательного паралича и синдрома паркинсонизма возникают в результате 586
перенесенных острых и хронических инфекций нервной системы (эпидемический энцефалит Экономо, клещевой, вирусный и другие виды энцефалитов). Причинами болезни могут служить церебральный атеросклероз, сосудистые заболевания головного мозга, опухоли, травмы нервной системы, длительное использование препаратов фенотиазинового ряда (аминазин, трифтазин), производных раувольфии, метилдофа — лекарственный паркинсонизм. Паркинсонизм может развиваться при острой или хронической интоксикации окисью углерода и марганца. В возникновении акинетико-ригидного синдрома может иметь значение наследственно обусловленное нарушение обмена катехоламинов в мозге или неполноценность ферментных систем, контролирующих этот обмен. Часто выявляется семейный характер заболевания при аутосомно-доминантном типе наследования. Подобные случаи относят к болезни Паркинсона. Различные экзо- и эндогенные факторы (атеросклероз, инфекции, интоксикации, травмы) способствуют проявлению генуинных дефектов в механизмах обмена катехоламинов в подкорковых ядрах и возникновению заболевания.
Основным патогенетическим звеном дрожательного паралича и синдрома паркинсонизма является нарушение обмена катехоламинов (дофамина, норадреналина) в экстрапирамидной системе. Дофамин выполняет самостоятельную медиаторную функцию в реализации двигательных актов. В норме концентрация дофамина в базальных узлах во много раз превышает его содержание в других структурах нервной системы. Ацетилхолин является медиатором возбуждения между полосатым телом, бледным шаром и черным веществом. Дофамин является его антагонистом, действуя тормо-зяще. При поражении черного вещества и бледного шара снижается уровень дофамина в хвостатом ядре и скорлупе, нарушается соотношение между дофамином и норадреналином, возникает расстройство функций экстрапирамидной системы. В норме импульсация модулируется в сторону подавления хвостатого ядра, скорлупы, черного вещества и стимулирования бледного шара. При выключении функции черного вещества возникает блокада импульсов, поступающих из экстрапирамидных зон коры большого мозга и полосатого тела к передним рогам спинного мозга. В то же время к клеткам передних рогов поступают патологические импульсы из бледного шара и черного вещества. В результате усиливается циркуляция импульсов в системе альфа- и гамма-мотонейронов спинного мозга с преобладанием альфа-активности, что приводит к возникновению паллидарно-нигральной ригидности мышечных волокон и тремора — основных признаков паркинсонизма.
Патоморфология. Основные патологоанатомические изменения при паркинсонизме наблюдаются в черном веществе и бледном шаре в виде дегенеративных изменений и гибели нервных клеток. На месте погибших клеток возникают очаги разрастания глиальных элементов или остаются пустоты.
Клинические проявления. Основной клинический синдром — акинетико-ригидный или гипертонически-гипокинетический. Для Дрожательного паралича и паркинсонизма характерны гипо- и акинезия. Появляется своеобразная сгибательная поза: голова и туловище наклонены вперед, руки полусогнуты в локтевых, лучезапястных и фаланговых суставах, нередко плотно приведены к боковым поверхностям грудной клетки. тУловища, ноги полусогнуты в коленных суставах. Отмечается бедность мимики. Темп произвольных движений с развитием заболевания постепенно
может наступить полная обездвижен-
ишедляется, иногда довольно Ра"° ^ шаркающими шагами. Нередко
ность. Походка характеризуется «да i P (пропульсии).
наблюдается склонность к непроизвольному ^ ^ как бы <<дап>
Если толкнуть больного вперед он ™ ведет к бегу „азад (ретро-
няя свой центр ^"лГтаро^льсш.) Движения наблюдаются также пульсии), в ст°Р°ьну<^тТьаРЗшуть голову назад. Часто при резко выра-при попытке сесть, к™'™ну ^ш каталептические. Акинез и женном синдроме позы больного Н^ПО"И"^ТВЛЯ1ОТСЯ в мускулатуре лица, Г^еская «SS^SSS^SSSSSSm При ходьбе or-
шечн^Гсопрот—ие вследствие повышения тонуса мышц-антагонистов, Аеномен «Жтого колеса» (возникает впечатление, что суставная поверх-itcTb сосгоиг L сцепления двух зубчатых колес). Повышение тонуса мышц-антагонистов при пассивных движениях можно определить следующим приемом: если поднять голову лежащего, а потом резко отпустить руку то голова не упадет на подушку, а опустится относительно плавно. Иногда голова в положении лежа несколько приподнята - феномен «воображаемой подушки».
Тремор — характерный, хотя и не обязательный для синдрома паркинсонизма симптом. Это ритмичное, регулярное, непроизвольное дрожание конечностей, лицевой мускулатуры, головы, нижней челюсти, языка, более выраженное в покое, уменьшающееся при активных движениях. Частота колебаний 4—8 в секунду. Иногда отмечаются движения пальцами в виде «скатывания пилюль», «счета монет». Тремор усиливается при волнениях, практически исчезает во сне.
Психические нарушения проявляются утратой инициативы, активности, сужением кругозора и интересов, резким понижением различных эмоциональных реакций и аффектов, а также некоторой поверхностью и медлительностью мышления (брадифрения). Наблюдаются брадипсихия — трудное активное переключение с одной мысли на другую, акайрия — прилипчивость, вязкость, эгоцентризм. Иногда возникают пароксизмы психического возбуждения.
Вегетативные нарушения проявляются в виде сальности кожи лица и волосистой части головы, себореи, гиперсаливации, гипергидроза, трофических нарушений в дистальных отделах конечностей. Выявляется нарушение постуральных рефлексов. Иногда специальными методами исследования определяется нерегулярное по частоте и глубине дыхание. Сухожильные рефлексы, как правило, без отклонений. При атеросклеротическом и постэнцефалитическом паркинсонизме могут определяться повышение сухожильных рефлексов и другие признаки пирамидной недостаточности. При постэнцефалитическом паркинсонизме встречаются так называемые окулогирные кризы — фиксация взора кверху в течение нескольких минут или часов; иногда голова при этом запрокинута. Кризы могут сочетаться с нарушением конвергенции и аккомодации (прогрессирующий супранукле-арный паралич).
Принято различать несколько клинических форм дрожательного паралича и паркинсонизма: ригидно-брадикинетическую, дрожательно-ригид-ную и дрожательную. Ригидно-брадикинетичвская форма характеризуется
повышением тонуса мышц по пластическому типу, прогрессируют, медлением активных движений вплоть до обездвиженности; появляются мышечные контрактуры, флексорная поза больных. Эта форма паркинсонизма, наиболее неблагоприятная по течению, чаще наблюдается при атеросклеротическом и реже при постэнцефалитическом паркинсонизме. Лро-жательно-ригидная форма характеризуется тремором конечностей, преимущественно их дистальных отделов, к которому с развитием заболевания присоединяется скованность произвольных движений. Для дрожательной формы паркинсонизма характерно наличие постоянного или почти постоянного средне- и крупноамплитудного тремора конечностей, языка, головы, нижней челюсти. Тонус мышц нормальный или несколько повышен. Темп произвольных движений сохранен. Эта форма чаше встречается при постэнцефалитическом и посттравматическом паркинсонизме.
Данные лабораторных и функциональных исследований. При посттравматическом паркинсонизме выявляется повышение давления цереброспинальной жидкости при нормальном клеточном и белковом ее составе. При паркинсонизме, возникающем вследствие отравления окисью углерода, в крови обнаруживается карбоксигемоглобин, при марганцевом паркинсонизме — следы марганца в крови, моче, цереброспинальной жидкости. Глобальная электромиография при дрожательном параличе и паркинсонизме выявляет нарушение электрогенеза мышц — повышение биоэлектрической активности мышц в покое и наличие ритмических групповых разрядов потенциалов. При электроэнцефалографии обнаруживаются преимущественно диффузные негрубые изменения биоэлектрической активности головного мозга.
Диагностика и дифференциальный диагноз. В первую очередь следует дифференцировать болезнь Паркинсона от синдрома паркинсонизма. Для постэнцефалитического паркинсонизма характерны глазодвигательные симптомы; могут наблюдаться кривошея, явления торсионной дистонии, которые никогда не наблюдаются при дрожательном параличе. Встречаются нарушения сна, дыхательные дискинезии с приступами зевоты, кашля, адипозогенитальные нарушения, вегетативные пароксизмы. Посттравматический паркинсонизм достоверно можно диагностировать у больных молодого и среднего возраста. Заболевание развивается после тяжелой, иногда повторной черепно-мозговой травмы. Для посттравматического паркинсонизма нехарактерны антеретропульсии, судорога взора, расстройства жевания, глотания, дыхания, каталептоидные явления. В то же время часто встречаются вестибулярные расстройства, нарушение интеллекта и памяти, зрительные галлюцинации (вследствие поражения коры большого мозга). Нередко отмечаются регредиентное течение или стабилизация патологического процесса. Для диагностики марганцевого паркинсонизма имеют значение анамнез (сведения о работе в контакте с марганцем или его окислами), обнаружение марганца в биологических жидкостях. Диагностика оксиугле-родного паркинсонизма базируется на определении в крови карбоксигемо-глобина.
При атеросклеротическом паркинсонизме дрожание и ригидность сочетаются с признаками церебрального атеросклероза или возникают после острых нарушений мозгового кровообращения. Выявляются очаговые неврологические симптомы в виде пирамидной недостаточности, выраженных псевдобульбарных симптомов. Часто определяется унилатеральность ригидности и скованности. В крови обнаруживается дисяипидемия, характер-
пая для атеросклероза. Регистрируются определенные изменения РЭГ в
виде уплощения пульсовых вол"- болезнь Паркинсона, может на-
гпессиоуюшие явления мозжечковой атаксии.
грессируюши ^ ^ ^ 03 заболевание неуклонно прогрессирует.
Исключение составляют некоторые формы, обусловленные лекарственными интоксикациями (при отмене препаратов может наступить улучшение состояния). Общепризнано, что лечение в начальной стадии позволяет уменьшить выраженность симптомов, замедлить прогрессирование заболевания В поздних стадиях лечебные мероприятия менее эффективны. Заболевание приводит к инвалидизации в течение нескольких лет. Даже лечение леводопой в настоящее время замедляет течение на непродолжительное время. Это подтверждает положение, что в основе заболевания лежит не только первичный биохимический дефект, но и еще не изученный нейро-патологический процесс.
Лечение. Лечение больных с дрожательным параличом и синдромом паркинсонизма должно быть комплексным, длительным и включать специфические антипаркинсонические препараты, седативные средства, физиотерапевтические процедуры, лечебную физкультуру, психотерапию с учетом этиологического фактора, возраста больных, клинической формы и стадии болезни, а также наличия сопутствующих заболеваний. При легких формах вначале назначают амантадин (мидантан) и парасимпатолитики, так как они вызывают меньше побочных явлений. Применяют центральные парасимпатолитики (циклодол, наркопан), пиридоксин, амантадин, агонисты дофаминовых рецепторов (бромокриптин, лизурид).
При выраженных клинических проявлениях паркинсонизма в настоящее время базисным препаратом является леводопа, обычно в сочетании с ингибитором декарбоксилазы. Дозы увеличивают медленно, в течение нескольких недель, до получения клинического эффекта. Побочные действия препарата — дистонические нарушения и психозы. Леводопа, попадая в ЦНС, декарбоксилируется в допамин, необходимый для нормальной функции базальных ганглиев. Препарат влияет прежде всего на акинезию и в меньшей степени - на другие симптомы. При сочетании леводопы с ингибитором декарбоксилазы можно уменьшить дозу леводопы и тем самым уменьшить риск развития побочных явлений.
В арсенале симптоматических антипаркинсонических средств большое место занимают холинолитические препараты, которые, блокируя м- и н-холинорецепторы, способствуют расслаблению поперечнополосатой и гладкой мускулатуры, уменьшают насильственные движения и явления брадикинеэии. Это естественные и синтетические атропиноподобные препараты: беллазон (ромпаркин), норакин, комбипарк. Применяют также препараты фенотиазинового ряда: динезин, депаркол, парсидол дипразин. Основная причина многообразия медикаментозных препаратов, используе-590
мых для лечения паркинсонизма, в недостаточной их лечебной эффективности, наличии побочных явлений, индивидуальной непереносимости и быстром привыкании к ним.
Хирургическое лечение. Несмотря на большие успехи, достигнутые в медикаментозном лечении паркинсонизма, возможности его в ряде случаев ограничены.
Наиболее широко применяемый препарат леводопа в большей степени способствует устранению таких симптомов болезни, как акинезия, обшая скованность, в меньшей степени он влияет на ригидность мышц и тремор. Приблизительно у 25 % больных этот препарат практически неэффективен или плохо переносится.
В этих случаях возникают показания для стереотаксической операции на подкорковых узлах. Обычно производится локальное разрушение вент-ролатерального ядра зрительного бугра, субталамических структур или бледного шара.
С помощью операции удается в большинстве случаев добиться положительного эффекта — снижения мышечного тонуса, ослабления или прекращения тремора, уменьшения гипокинезии.
Операция обычно выполняется на стороне, противоположной той, на которой преобладают симптомы паркинсонизма. При показаниях производится двустороннее разрушение подкорковых структур.
В последние годы для лечения паркинсонизма используется также имплантация эмбриональной ткани надпочечника в полосатое тело. О клинической эффективности таких операций пока говорить преждевременно.
Стереотаксические операции на подкорковых структурах применяются также и при других заболеваниях, сопровождающихся насильственными движениями (гемибаллизм, хореоатетоз, кривошея и некоторые другие).
Трудоспособность при болезни Паркинсона и паркинсонизме зависит от степени выраженности двигательных нарушений, вида профессиональной деятельности. При легких и умеренных нарушениях двигательных функций больные длительно сохраняют трудоспособность при различных видах умственного труда, а также работах, не связанных с физическим напряжением и выполнением точных и координированных движений. При выраженных проявлениях заболевания больные нетрудоспособны и нуждаются в посторонней помощи.
24.2.3. Гепатоцеребральная дистрофия
Гепатоцеребральная дистрофия (гепатолентикулярная дегенерация, болезнь Вестфаля—Вильсона—Коновалова) — хроническое прогрессирующее наследственно-дегенеративное заболевание, характеризующееся сочетан-ным поражением подкорковых узлов ЦНС и печени. Описано в 18S К. Вестфалем и в 1912 г. С. Вильсоном. Термин «гепатоцеребральная дистрофия» предложен Н.В. Коноваловым.
Этиология и патогенез. Заболевание наследственное. Тип наследования аутосомно-рецессивный. Ведущим патогенетическим звеном является генетически обусловленное нарушение синтеза белка и РУлоплазмина, входящего в состав а2-глобулина, транспортирующего медь Вследствие этого создается высокая концентрация меди в крови
происходит ее отложение в органах и тканях, W™
мозге, роговице, а также в почках и других органах.
меди связано с блоком сульфгидрильных ФУпп в
тах, что приводит к нарушению окислительно-во
сов в клетке в мозге, печени, почках, селезенке, рогови-
^ оболочке, хрусталике глаза определяются дегенеративные из- выраженные в подкорковых ядрах. Обнаруживаются так^ист^еск2е изменения нервных клеток, очаговые размягчения "вой ™ с образованием микрокист, разрастанием глии. Выявляют-я измененГмелких сосудов мозговой ткани, кровоизлияния вокруг них, периваскулярный отек. Постоянным признаком заболевания является цир-
3 ТлТн ические проявления. Складываются из симптомов поражения ЦНС и внутренних органов. У больных появляются и нарастают мышечная ригидность, разнообразные гиперкинезы, псевдобульбарные симптомы, прогрессирующее снижение интеллекта, нарушения функции печени и изменение радужной оболочки. Ведущим является синдром экстрапирамидных расстройств: ригидность мышц туловища, конечностей, лица, глотки и как следствие этого — нарушения походки, глотания, речи. Параллельно возникают гиперкинезы различного характера: тремор, атетоз, торсионная дистония, интенционное дрожание, усиливающиеся при попытке выполнения произвольных движений. Гиперкинезы имеют неритмичный характер.
В зависимости от выраженности и сочетания клинических проявлений, возраста, в котором возникло заболевание, и степени поражения печени выделяют четыре формы гепатоцеребральной дистрофии.
1. Ранняя ригидно-аритмогиперкинетическая форма имеет наиболее
злокачественное течение. Неврологические проявления развиваются в воз
расте 7—15 лет. Этому, как правило, предшествуют признаки поражения
печени. В клинической картине преобладают мышечная ригидность и ги
перкинезы.
2. Дрожательно-ригидная и дрожательная формы, проявляющиеся в
более позднем возрасте (17—20 лет). Характеризуются одновременным по
явлением ригидности и дрожания, которое часто бывает первым признаком
заболевания; постепенно нарастая, оно может становиться общим, захва
тывая мышцы туловища, конечностей, лица, челюстей, мягкого неба, над
гортанника, голосовых связок, дыхательную мускулатуру, диафрагму. Нару
шается глотание, речь становится скандированной. Часто отмечаются вы
раженные изменения психики.
. Экстрапирамидно-корковая форма, выделенная Н.В. Коноваловым, отличается расстройством высших мозговых функций, наличием параличей, часто эпилептических припадков, грубым снижением интеллекта с изменениями личности.
4. Абдоминальная форма характеризуется преимущественным нарушением функции печени. Неврологические симптомы присоединяются в более поздних стадиях болезни.
Течение и прогноз. Течение неуклонно прогрессирующее. Продолжительность жизни зависит от клинической формы заболевания, своевременности начатого лечения. Средняя продолжительность жизни больных без лечения около 6 лет.
Данные лабораторных и функциональных исследований. В сыворотке крови обнаруживаются значительное снижение содержания церулоплазмина (ниже 10 ЕД при норме 25—45 ЕД), гипопро-теинемия, гиперкупрурия (до 1000 мкг/сут и выше при норме 150 мкг/сут) и гипераминоацидурия (до 1000 мг/сут при норме 350 мг/сут). Возможны также повышение содержания аммиака в крови, изменение печеночных проб. Патогномоничным признаком гепатоцеребральной дистрофии является роговичное кольцо Кайзера—Флейшера, которое представляет собой отложение пигмента, содержащего медь, по периферии радужной оболочки.
Дифференциальный диагноз. Заболевание следует дифференцировать от летаргического энцефалита, малой хореи, дегенеративных подкорковых заболеваний, рассеянного склероза. Выявление семейного анамнеза, характерной клинической картины, роговичного кольца Кайзера—Флейшера, низкого уровня церулоплазмина в крови и повышения экскреции меди с мочой у больных и их родственников позволяет поставить диагноз гепатоцеребральной дистрофии.
Лечение. Основной целью лечения является выведение из организма избытка меди. Для этого используют тиоловые препараты, к которым относятся унитиол, декаптол и D-пеницилламин. Дозы подбираются индивидуально. D-пеницилламин в среднем назначают в дозе от 0,45 до 2 г в сутки после еды. Препарат необходимо принимать в течение всей жизни. Наиболее эффективно лечение в ранних стадиях болезни. Унитиол назначают повторными курсами по 5 мл 5 % раствора внутримышечно ежедневно или через день (на курс 25 инъекций с перерывом между курсами 5—6 мес). Лечение сочетают с препаратами, улучшающими функции печени. Рекомендуется специальная диета с ограничением продуктов, богатых медью (печень, грибы, шоколад, устрицы и др.), животных жиров, белков. Пища должна быть богата витаминами и углеводами.
24.2,4. Торсионная дистония •
Хроническое прогрессирующее заболевание нервной системы, клинически проявляющееся изменениями мышечного тонуса и непроизвольными тоническими сокращениями мышц туловища и конечностей.
Этиология и патогенез. Различают идиопатическую (семейную) торсионную и симптоматическую дистонию. Тип наследования при идиопатической торсионной дистонии как аутосомно-доминантный, так и аутосомно-рецессивный. Симптоматическая торсионная дистония встречается при гепатоцеребральной дистрофии, хорее Гентингтона, опухолях мозга, эпидемическом энцефалите, детском церебральном параличе. Имеются указания, что в патогенезе наследственной торсионной дистонии имеет значение нарушение допаминового обмена. При обследовании у эг больных обнаруживается повышение содержания допамин-р-гидроксилазы в сыворотке крови.
Патоморфология. Дистрофические изменения обнаруживаются преимущественно в мелких нейронах в области скорлупы чечевицеоо-Разного ядра, реже — в других базальных ганглиях.
Клинические проявления. Развивается заболевание посте пенно, в Ь случаев в возрасте до 15 лет. В детском возрасте первыми симп-
S93
I n
томами болезни могут быть нарушение походки, спастическая кривошея; у юмТы^ащГ1^2?а1агся первично-генерализованные формы. В резуль-тГ нарушения соотношения функции мышц-синергистов и антагонистов возникатнасильственные длительные тонические сокращения мышц ту-Гвиш^овГ^азовоп, пояса, конечностей, обычно ротаторного характера сочетающиеся с атетоидными движениями в пальцах. Создается впечатление чтТмышцы постоянно сокращаются для преодоления действия антагонистов Возникающие позы, даже самые неудобные, сохраняются в Учение длительного времени. Гиперкинеэы усиливаются при волнении, активных движениях, во сне исчезают. Постепенно, по мере прогрессиро-вания заболевания, поза пациента становится постоянно дистоническои, с усиленным поясничным лордозом, флексией бедер, медиальной ротацией рук и ног В зависимости от распространенности дистонических явлений выделяют локальную и генерализованную формы заболевания. При локаль^ ньгх дистонических симптомах возникает тоническое сокращение отдельных мышечных групп, нарушаются произвольные движения и возникает аномальная поза. К таким симптомам относятся спастическая кривошея, писчий спазм, оромандибулярная дистония (открывание и закрывание рта и непроизвольные движения языка), блефароспазм, щечно-лицевая, щечно-язычная дистония, хореоатетоз.
Течение и прогноз. Заболевание в большинстве случаев неуклонно прогрессирует. Иногда отмечаются различной длительности ремиссии. Быстро происходит глубокая инвалидизация больных и наступает летальный исход, особенно при генерализованной форме.
Лечение. Длительное, симптоматическое. Применяют комбинации холинолитиков и седативных препаратов, в некоторых случаях эффективно использование леводопы. Назначается также галоперидол или резерпин. Очень редко прибегают к стереотаксическим операциям на подкорковых ядрах.
24.2.5. Хорея Гентингтона -
Хроническое прогрессирующее наследственно-дегенеративное заболевание, характеризующееся нарастающим хореическим гиперкинезом и де-менцией. Описано Дж.Гентингтоном в 1872 г. Частота хореи Гентингтона от 2 до 7 случаев на 100 000 населения. Применяется также термин «демен-ция хореическая».
Этиология и патогенез. Заболевание наследственное. Тип наследования аутосомно-доминантный с высокой пенетрантностью (80-го %). Мужчины болеют чаще. Патогенез изучен недостаточно В клетках головного мозга в ряде случаев обнаружен недостаток ГАМ К в клетках черного вещества - повышение содержания железа, имеются нарушения допаминового обмена. Изучен патофизиологический механизм двигательных расстройств. Блок стрионигральных связей обусловливает отсутствие контроля над содружественностью движений и мышечного тонуса со стороны черного вещества, которое передает полученные от премоторной зоны коры импульсы к клеткам передних рогов спинного мозга в нерегулярной последовательности.
Патоморфология. Обнаруживается атрофия мозга В подкорковых ганглиях, преимущественно в скорлупе и хвостатом ядре, определя-

Рис. 24.4. Хорея Гентингтона (а—в).
ются грубые дегенеративные изменения мелких и крупных клеток, уменьшение их числа, разрастание глиальных элементов.
Клинические проявления. Возникает заболевание обычно в возрасте 30 лет и старше. Первыми симптомами могут быть интеллектуальные расстройства, в дальнейшем постепенно развивается деменция. Одновременно появляются хореические гиперкинезы: быстрые, неритмичные, беспорядочные движения в различных мышечных группах. Выполнение произвольных движений затруднено вследствие гиперкинезов и сопровождается рядом ненужных движений. Так, например, при ходьбе больные гримасничают, жестикулируют, приседают, широко расставляют руки (рис. 24.4). Однако даже при выраженном гиперкинезе, особенно в начале болезни, они могут его сознательно подавлять. Речь затруднена и также сопровождается излишними движениями. Мышечный тонус снижен. Парезы конечностей и другие очаговые неврологические симптомы не определяются. Нередко наблюдаются эндокринные и нейротрофические расстройства. В 5—16 % случаев диагностируется атипичный акинетико-ригидный вариант хореи Гентингтона. При этом развивается акинетико-ригидный синдром в сочетании с прогрессирующей интеллектуальной деградацией и умеренно выраженным хореическим гиперкинезом. Из насильственных движений преобладает хореоатетоз.
Течение. Заболевание неуклонно прогрессирует. Длительность егс 5—10 лет с момента возникновения первых симптомов. Более доброкачественное течение отмечается при атипичной акинетико-ригидной форме.
Данные лабораторных и функциональных и сел Д о в а н и й. На ЭЭГ отмечаются диффузные изменения биоэлектрической активности мозга. При компьютерной и МР-томографии выявляются расширение желудочков и так называемое вдавление таламуса, если забалева ние связано с поражением его мелких клеток, обнаруживаются признаки атрофии коры большого мозга. Имеются указания на возможность ранней.
S95
чув"
диагностические критерии. Г/лит„иггпиа СЛедует от хореического син-
Дифференцировать хорею Гентингтона следую и £ эннеАя
а возникающего при опухолях головною Mu^i a, v V *г
1в сосудистых заболеваний, а также старческой (сенильнои) хореи. Л е че ни е Для подавления гиперкинеза назначают антагонисты долами! Это препараты фенотиазинового ряда - трифтазин (7,5-10 мг в Г„в сочетании с транквилизаторами, допегитом, резерпином. Попытки лечения больных, страдающих хореей Гентингтона, с помощью стереотак-сических операций оказались безуспешными.
24.2.6. Болезнь Фридрейха *
Семейная атаксия Фрндрейха — наследственное дегенеративное заболевание нервной системы, характеризующееся синдромом поражения задних и боковых канатиков спинного мозга. Тип наследования аутосомно-рецес-сивный, с неполной пенетрантностью патологического гена. Мужчины и женщины болеют одинаково часто.
Патоморфология. Обнаруживаются дегенеративные изменения в проводящих путях задних и боковых канатиков спинного мозга, преимущественно пучков Голля, в меньшей степени — Бурдаха, Флексига, Го-верса, волокнах пирамидного пути, задних корешках, а также в клетках коры мозжечка, подкорковых ганглиев, коры большого мозга.
|
Клинические проявления. Начало заболевания относится к 6—15-летнему возрасту. Первым симптомом болезни является неустойчивая походка, которая была охарактеризована Шарко как табетически-мозжечковая. В ранних стадиях атаксия выражена преимущественно в ногах. По мере прогрессирования заболевания нарушения координации распространяются на верхние конечности и лицо. При неврологическом обследовании выявляются крупноразмашистый нистагм, атаксия в руках и ногах, адиадохо-кинез, дисметрия, скандированная речь, расстройства мышечно-суставного чувства и вибрационной чувствительности. Меняется почерк. Ранним симптомом является снижение, а затем угасание сухожильных и периостальных рефлексов. Мышечный тонус понижен. В более поздних стадиях болезни присоединяются афферентный парез нижних, а затем верхних конечностей, нередки патологические пирамидные рефлексы, дистальные мышечные атрофии. Интеллект сни-Рис. 24.5. Стопа Фрндрейха. жен.
Заболевание медленно прогрессирует. Средняя продолжительна жизни 10—15 лет с момента его развития.
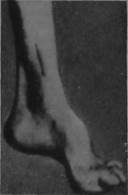
Диагностика и дифференциальный диагноз. Заболевание распознается на основании характерных симптомов — деформаций стоп (рис. 24.5) по типу стопы Фридрейха (высокий свод, экстензия основных фаланг пальцев стопы и флексия концевых фаланг), поражения миокарда, эндокринных расстройств.
Дифференцировать заболевание следует от церебрального сифилиса, рассеянного склероза, фуникулярного миелоза и других форм мозжечковых дегенерации.
Лечение. Применяются симптоматические средства: общеукрепляющие препараты, лечебная физкультура, массаж. В некоторых случаях производится хирургическая коррекция деформации стоп.
24.2.7. Наследственная мозжечковая атаксия Пьера Мари
Мозжечковая атаксия Пьера Мари — наследственное дегенеративное заболевание с преимущественным поражением мозжечка и его проводящих путей. Тип наследования аутосомно-доминантный. Возникает заболевание в возрасте 20 лет и старше.
Патоморфология. Выявляется дегенеративное поражение клеток коры и ядер мозжечка, спиноцеребеллярных путей в боковых канатиках спинного мозга, в ядрах моста и продолговатого мозга.
Клинические проявления. Заболевание проявляется нарушениями функций мозжечка и его связей. Наблюдаются атаксия при выполнении координаторных проб, нарушение походки, скандированная речь, интенционное дрожание, нистагм. Мозжечковые симптомы сочетаются с умеренными или выраженными признаками пирамидной недостаточности (повышение сухожильных и периостальных рефлексов, клонусы стоп), а иногда с глазодвигательными нарушениями (косоглазие, птоз, недостаточность конвергенции). Характерным признаком является в различной степени выраженное снижение интеллекта.
Диагностика и дифференциальный диагноз. Наибольшие трудности возникают при дифференциации наследственной мозжечковой атаксии Пьера Мари и атаксии Фридрейха. Нужно учитывать тип наследования заболевания, возраст, в котором развиваются первые симптомы, характер изменения сухожильных рефлексов (при атаксии Фридрейха они снижены), наличие зрительных и глазодвигательных расстройств при атаксии Пьера Мари, деформации стоп и скелета. Рассеянный склероз в отличие от семейной атаксии Пьера Мари характеризуется ремиттирукшшм течением, большей выраженностью нижнего спастического парапареза, расстройством функций тазовых органов.
Лечение. Симптоматическое.
24.2.8. Оливопонтоцеребеллярные дегенерации
Группа наследственных заболеваний нервной системы, характеризующихся
Дегенеративными изменениями нейронов мозжечка, ядер нижних олив;(Моста мозга, в ряде случаев — ядер черепных нервов каудальной группы, в
| Глава 25 |
иеньше* степени - g£ ^X
рогов спинного мозга, fj™ ^"^инических симптомов. По клас
^^bZl^^ 5 типов ояквопоитоцеребел-
лярных деднер^ йии^иця Л*****. Наследуется по
мышечная гипотония, дизартрии, интенционное дрожание), нервов (дизартрия, дисфагия), подкорковых; реже выявляются пирамидные и глазодвигатель-
ные с^^\теопонто1<ере6мярная дегенерация Фиклера-Винклера. Наследуется по аутосомно-рецессивному типу. Проявляется в возрасте от 20 до 80 лет симптомами поражения мозжечка, преимущественно атаксией в конечностях. Чувствительность и сухожильные рефлексы не изменены. Парезов не наблюдается.
Тип т — оливопонтоцеребелгярная дегенерация с ретинальнои бегенера- цией. Наследуется по аутосомно-доминантному типу. Возникает в молодом возрасте. Наряду с мозжечковыми и экстрапирамидными симптомами определяется прогрессирующее снижение остроты зрения вследствие пигментной дегенерации ганглиозных клеток сетчатки.
Тип IV — оливопонтоцеребеллярная дегенерацця Шута—Хайкмана. Наследуется по аутосомно-доминантному типу. Проявляется в детском и молодом возрасте. Кроме мозжечковых симптомов, выявляется поражение ядер VII, IX, X и XII пар черепных нервов (паралич лицевого нерва, буль-барные симптомы) и задних канатиков спинного мозга (расстройства мы-шечно-суставного чувства и вибрационной чувствительности).
Тип V — оливопонтоцеребеллярная дегенерация с деменцией, офтальмоплегией и экстрапирамидными нарушениями. Тип наследования аутосомно-доминантный. Развивается в среднем возрасте. Характеризуется деменцией, прогрессирующей офтальмоплегией, экстрапирамидными и мозжечковыми симптомами.
Дифференцировать оливопонтоцеребеллярные дегенерации следует от
наследственной атаксии Фридрейха и Пьера Мари, прогрессирующих форм
рассеянного склероза, опухолей мозжечка, ювенильных форм паркинсо
низма. г
Лечение. Симптоматическое. Проводят курсы неспецифического общеукрепляющего лечения, массаж, лечебную физкультуру.
МИАСТЕНИЯ
Миастения, астенический бульбарный паралич (nryasthenia gravis pseudoparalitica) характеризуется выраженной слабостью и утомляемостью мышц. При этом заболевании поражаются холинорецепторы постсинапти-ческих мембран. В процесс может вовлекаться любая мышца тела, однако имеется тенденция к преимущественному поражению мыши лица, губ, глаз, языка, глотки и шеи.
Этиология. Окончательно не выяснена. Возможны семейные случаи, но наследственный характер заболевания не доказан. Нередко имеется сочетание миастении с гиперплазией или опухолью вилочковой железы. Иногда наблюдаются миастенические синдромы при органических заболеваниях нервной системы (боковой амиотрофический склероз и др.), поли-и дерматомиозите, а также раке легкого, молочной железы, яичника, предстательной железы. Женщины заболевают чаще, чем мужчины. Наиболее часто болезнь начинается в возрасте 20—30 лет.
Патогенез. Установлено, что миастения — аутоиммунное заболевание, поскольку множественные аутоантитела, антитела к рецепторам постсинаптической мембраны нервно-мышечного синапса найдены в сыворотке таких больных. Миастения обусловлена образованием антител к рецепторам постсинаптической мембраны с деструкцией ее и блоком нервно-мышечной передачи.
Патоморфология. Не выявлено каких-либо постоянных специфических изменений в ЦНС, периферических нервах или мышцах. Иногда находят увеличение или опухоль вилочковой железы. В поперечнополосатых мышцах обнаруживают атрофические и дистрофические изменения отдельных волокон и инфильтрацию лимфогистиоцитарными элементами интерстициальной ткани.
Клинические проявления. Обычно проявляется утомляемостью мышц с сопутствующей слабостью, особенно глазных и мышц, ин-нервируемых бульбарными нервами. Слабость глазных мышц приводит к диплопии и косоглазию, одно- или двустороннему птозу, наиболее выраженному к концу дня. Нередко отмечается слабость лицевой и жевательной мускулатуры. Трудности речи и глотания могут быть выявлены после более или менее длительного разговора и приема пищи. Возможны слабость и утомляемость мышц языка и носовой оттенок голоса. Может быть поражена и другая поперечнополосатая мускулатура конечностей и шеи, что приводит к генерализованной слабости. Определяется истощаемость сухожильных рефлексов. При повторной электрической стимуляции выявляются патологическая утомляемость мышц, выраженная способность к восстаноаче-нию после короткого отдыха. Характерны лабильность, динамичность симптомов с их усилением при чтении, фиксации взгляда, иногда общей физической нагрузке. Миастения может быть генерализованной и лохааь-
| „а* ггтптки гортани мимической мускулатуры или Гет с^~ся |
| зоды (короткие спонтанные ния на протяжении наступить резкое ухудшение мышечной слабостью лаз (афония, Дизартрия |
^^ проявле-
* " "™™"™ срока) У больных миастенией может значительН°Г^^ де криза с генерализованной
^ дУыхаНия, психомоторным головного мозга с расстрой-
хтг^ ^~
астения диагностируется на основании жалоб на утомляемость, усиление имеющихсярасстройств к вечеру и при физической нагрузке. Важное зна-чениеТмеет прозериновая проба: резкое уменьшение симптомов через 30- 60 мин после введения 1-2 мл 0,05 % раствора прозерина подкожно. Типично изменение электровозбудимости мышц: быстрое истощение их сокращения при повторных раздражениях током с восстановлением возбудимости после отдыха. Весьма ценным методом в диагностике миастении является электромиографическое исследование. При стимуляционной электромиографии регистрируется нормальный суммарный вызванный потенциал действия мышцы, амплитуда которого уменьшается при ритмической стимуляции частотой 3—5 и 50 импульсов в 1 с.
Дифференциальный диагноз проводится со стволовым энцефалитом, опухолью ствола мозга, базальным менингитом, глазной формой миопатии, полимиозитом, нарушением мозгового кровообращения в вертебробази-лярной системе.
Лечение. Направлено на коррекцию относительного дефицита ацетилхолина и подавление аутоиммунного процесса. С целью компенсации расстройств нервно-мышечной передачи используют антихолинэсте-разные средства: прозерин, оксазил, пиридостигмина бромид (местинон, калимин, амиридин). Важен выбор оптимальной индивидуально компенсирующей дозы в зависимости от клинической формы, тяжести симптоматики, сопутствующих заболеваний, реакции на препарат. При глоточно-лице-вой и глазной формах миастении более эффективны пиридостигмина бромид, прозерин и оксазил. Дозы препаратов и интервалы приема индивидуальны. Назначают хлорид или оротат калия, верошпирон, эфедрин. В тяжелых случаях вводят прозерин парентерально (1,5—2 мл 0,05 % раствора внутримышечно) за 20—30 мин до приема пищи. Прием больших доз анти-холинэстеразных препаратов может привести к холинергическому кризу. Основными методами лечения этого криза являются отмена антихолинер-гических средств и повторное введение атропина (0,5 мл 0,1 % раствора внутривенно или подкожно).
При миастеническом кризе, возникающем в результате недостаточной дозы антихолинэстеразных средств, срочно вводят прозерин внутривенно (0,5—1 мл 0,05 % раствора) и внутримышечно (по 2—3 мл через 2—3 ч). Оксазил может быть введен в свечах. Применяют также 5 % раствор эфедрина подкожно, препараты калия внутривенно. Прогрессирующая и угрожающая жизни слабость дыхательных мышц может наблюдаться, несмотря на
введение больших количеств прозерина. Больным производят интубацию или трахеостомию, переводят на ИВЛ. Питание больных осуществляют через назогастральный зонд. Необходимо поддерживать баланс жидкости и электролитов, витаминов; по показаниям (метаболический ацидоз) вводится внутривенно капельно 1 % раствор бикарбоната натрия.
Основными методами патогенетического лечения больных миастенией являются тимэктомия, рентгенотерапия и гормональная терапия. Хирургический метод (тимэктомия) показан всем больным в возрасте до 60 лет. страдающим миастенией, но находящимся в удовлетворительном состоянии; Он абсолютно показан при опухоли вилочковой железы. Рентгенотерапия на область этой железы назначается после неполной тимэктомии, при глазной форме миастении, а также при наличии противопоказаний к операции у больных пожилого возраста с генерализованной формой миастении. В тяжелых случаях — при генерализованной миастении — показано лечение иммуносупрессивными препаратами. Назначают кортикостерои-ды, лучше всего метилпреднизолон (по 100 мг через день). Длительность приема максимальной дозы кортикостероидов ограничивается наступлением значительного улучшения, которое позволяет впоследствии снижать дозу до поддерживающей.
Прогноз. Возможны спонтанные ремиссии, но, как правило, наступает обострение. Беременность обычно вызывает улучшение, хотя наблюдается и усиление имеющихся расстройств. Возможны миастенические кризы с летальным исходом вследствие дыхательной недостаточности. После криза может быть ремиссия. Передозировка антихолинэстеразных препаратов может вызвать мышечную слабость, напоминающую миастени-ческий криз. Раннее применение интубации или трахеостомии в сочетании с ИВЛ позволяет снизить летальность при миастеническом кризе с острой дыхательной недостаточностью.

|
Глава 26
ФАКТОРОВ
давления, изменения потоотделения, нарушения цикличности сна и I ствования, аппетита, половой функции и др.
В зависимости от характера и особенностей воздействие ных факторов могут проявляться диффузные и очаговые орг. симптомы. При действии определенных факторов (вибрашг отморожения) поражается преимущественно периферическая нервная система.
К экстоемальным могут быть отнесены факторы, которые по интенсивности ил^ характеру воздТйствия стоят на крайних границах или за пределами 2зиолоГческих возможностей адаптационных реакции организма. Часть экстремальных факторов может действовать на организм извне: условия деятельности связанные со зрительным и слуховым напряжением, избыточной информацией или сенсорной изоляцией, физическим переутомлением и гипокинезией, кислородным голоданием, перегреванием или переохлаждением а также действие интоксикаций, проникающей радиации, СВЧ-поля, невесомости и др. Для ряда профессий экстремальность условии работы'определяется не столько характером внешних воздействий, сколько психоэмоциональной напряженностью, связанной с сознанием ответственности или жизненной значимости правильной оценки информации и готовности к адекватной реакции.
Экстремальными могут быть необычные условия жизни и труда со значительным и длительным сдвигом стереотипов биологических ритмов, объединяемых понятием «десинхроноз» (например, нарушение суточного ритма сна и бодрствования). Источником срыва могут быть трудности адаптации. Примером может служить работа операторов, летчиков, командиров и других специалистов надводных кораблей и подводных лодок. Психоэмоциональное перенапряжение может быть обусловлено также фактором непрерывности и строгой подчиненности порядка рабочих операций, необычностью и «избыточностью» информации при лимитированной по времени и способу выполнения психомоторной деятельности.
В зависимости от особенностей личности, резерва тех или иных функциональных систем и характера воздействия наблюдаются разнообразные клинические варианты неврологических расстройств, начиная от дезадап-тационных астенических симптомов, вегетативно-сосудистых дистоний и преходящих явлений межполушарной пирамидной асимметрии до органических неврологических синдромов с соответствующими морфологическими изменениями.
В зависимости от характера воздействия и особенностей личности:ловека могут преобладать явления невроза или астении, сочетание которых дает основание иногда говорить об астеноневротическом синдроме. < геническим явлениям обычно сопутствуют различные изменения функ-111111 вегетативных отделов нервной системы. Чаще всего это проявляется пивной лабильностью. Более выраженная общая и распространенная 1ИЯ - вегетативные дистоний, определяемые как нейроциркуляторная 1И< ГОНИЯ, при которых ведущими являются нарушения вегетативной регу- [ЯЦИИ функций сердечно-сосудистой системы. Проявления вегетативной пч ниши весьма разнообразны: колебания частоты пульса и артериального
26.1. Общее охлаждение
Патоморфология. Обнаруживаются распространенные изменения типа хроматолиза, реактивные поражения нервных волокон и концевых структур. Изменения отмечаются в коре полушарий большого мозга, мозжечка, подкорковых узлах, в спинном мозге. Обращает на себя внимание значительное поражение чувствительных узлов: спинномозговых (рис. 26.1), полулунного, а также многих вегетативных узлов.
У людей, выведенных из состояния глубокого охлаждения, наблюдаются обратимые функциональные нарушения: изменения нервно-рефлекторной деятельности, гипертермия, вегетативно-трофические расстройства (очаги облысения, трофические язвы), сдвиги в системе красной и белой крови (анемия, лейкоцитоз, возрастание СОЭ), сопутствующие инфекционные заболевания.
Клинические проявления. Симптомы острого охлаждения зависят от степени охлаждения, длительности пребывания на холоде и быстроты падения температуры тела. При остром охлаждении первоначально возникает возбуждение нервной системы, в частности аппаратов, регулирующих дыхание, кровообращение и температуру. Отмечаются учащение пульса, повышение артериального давления. Наблюдаются усиление двигательной активности, дрожание в мышцах туловища и конечностей, иногда фасцикулярные подергивания в мышцах или их судорожное сокращение. При этом благодаря усилению обмена некоторое время температура тела сохраняется в пределах нормы, но затем начинает постепенно падать. При температуре тела 34—31 °С происходит угнетение некоторых функций нервной системы, развиваются общая заторможенность, замедленность реакций на внешние раздражители, сонливость.
Различают три степени (формы) переохлаждения: легкую (адинамичес-кую), среднюю (ступорозную), тяжелую (коматозную). При адинамической форме охлаждения пострадавшие заторможены, жалуются на общую слабость, головную боль, головокружение. Иногда наблюдаются состояние благодушия, эйфория, снижение критики в оценке окружаюшей обстановки и своего состояния. Речь тихая и медленная. Зрачки обычной величины, одинаковые, их реакция на свет становится менее живой. Отмечаются адинамия, снижение тонуса мышц конечностей. Сухожильные рефлексы снижены, кожные — вялые. Пульс у большинства больных замедлен до 40—60 в минуту. Артериальное давление меняется в небольших пре Тоны сердца приглушены. Дыхание не нарушено. Ректальная темпер 30—32 °С. В таком состоянии пострадавшие могут сами добраться ао пункта, некоторые способны самостоятельно принимать пищу Про:,, тельность астенического состояния при выведении из охлаждения в этих случаях не превышает нескольких суток.
Рис. 26.1. Клетка спинномозгового узла при охлаждении. Электронограмма.
При ступорозном охлаждении отмечается выраженная брадикардия (около 40 в минуту). Пульс очень слабый, малого наполнения, аритмии отмечаются редко. Резкое похолодание конечностей. Тоны сердца приглушены Артериальное давление чаще понижено или изредка повышено. Дыхание ослаблено и замедлено (до 8-10 в минуту). Ректальная температура 29—31 °С. Пострадавший находится в состоянии ступора, не может самостоятельно передвигаться. Нередко развивается расстройство памяти, сходное с корсаковским синдромом, которое сочетается с благодушным настроением, эйфорией, выраженным снижением критики. При согревании отмечаются общая заторможенность, скованность движений. Зрачки чаще расширены, но иногда в течение часа можно наблюдать по нескольку Ра-смену их сужения и расширения. Параллельно сужению зрачков обычно усиливается тонус скелетной мускулатуры. Сухожильные рефлексы оживлены. При дальнейшем снижении температуры тела продолжает нарастать угнетение функции ЦНС, падает возбудимость коры головного мозга, воз можно недержание мочи и кала.
При последующем падении температуры тела (ниже 28 °С) развивается тяжелая форма охлаждения — коматозная. Пострадавшие постепенно утрачивают сознание, непроизвольно двигают головой и руками, временами приоткрывают глаза и бессознательно переводят вгляд из стороны в сторо ну. Зрачки сужены, на свет не реагируют. Корнеальный рефлекс ослаблен либо отсутствует. Могут отсутствовать сухожильные и кожные рефлексы-Отмечаются судорожные тонические сокращения мускулатуры лица и ко
нечностей с преобладанием тонуса во флексорных группах, изредка — тризм, напряжение мышц брюшного пресса. Нарушаются вазомоторные реакции. Пульс на лучевых артериях не прощупывается, на плечевых — едва ощутим. Тоны сердца приглушены. Пульсовое давление, как и при охлаждении средней тяжести, чаще понижено. Дыхание резко ослаблено, иногда типа Чейна—Стокса. Жизненные функции постепенно угасают, однако при своевременном и правильном лечении пострадавшие могут быть выведены даже из глубокого коматозного состояния, причем постепенно восстанавливаются и функции нервной системы.
У людей, длительно находившихся на холоде, иногда развивается миалги-ческая форма общего охлаждения. Они сохраняют вынужденное («согбенное») положение туловища и с трудом передвигаются. Определяются ограничение движений позвоночника вперед и назад, болезненность мышц, особенно длинных мышц спины, резкое повышение их механической возбудимости.
Своеобразной реакцией нервной системы на холодовую травму, встречающейся относительно редко, является холодовый паралич. Для этой формы типично появление вслед за охлаждением пареза конечности, сохраняющегося до суток и сопров
Дата публикования: 2014-11-18; Прочитано: 1723 | Нарушение авторского права страницы | Мы поможем в написании вашей работы!
