 |
Главная Случайная страница Контакты | Мы поможем в написании вашей работы! | |
Тема 6.Гемобластозы
|
|
Гемобластозы – группа опухолей, субстратом которых являются клетки кроветворной ткани.
По уровню заболеваемости гемобластозы занимают 5-6 место среди злокачественных новообразований. Заболеваемость мужчин выше, чем женщин при всех формах гемобластозов.
Классификация. Единой классификации нет. По клинической классификации гемобластозы делят на три группы опухолей:
1) лейкозы - злокачественные опухоли кроветворной ткани с первичной повсеместной локализацией в костном мозге;
2) гематосаркомы – внекостномозговые, первоначально локальные опухоли (преимущественно в лимфатических узлах) представленные разрастанием бластных клеток, образующих солидные опухоли с их возможной генерализацией в кроветворные органы;
3) лимфомы - опухоли, состоящие из зрелых лимфоцитов и образованные разрастанием ткани идентичной лимфатическому узлу. На основе клинической классификации гемобластозов совершенствуются классификационные характеристики групп и форм этих заболеваний.
Лейкозы, первичное опухолевое перерождение которых развивается в миелоидной или лимфоидной гемопоэтической клетке, по характеристике морфологического субстрата делят на острые и хронические формы. При острых лейкозах субстратом злокачественной опухоли являются молодые бластные клетки. В классификации острых лейкозов выделяют острые лимфобластные лейкозы (ОЛЛ) и острые миелобластные лейкозы (ОМЛ):
М0 — с недифференцированными бластными клетками;
М1 — острый миелобластный лейкоз без признаков созревания клеток;
М2 — острый миелобластный лейкоз с признаками созревания клеток;
М3 —промиелоцитарный лейкоз;
М4 — миеломоноцитарный лейкоз;
М5 — моноцитарный (монобластный) лейкоз;
М6 — острый эритролейкоз;
М7 — мегакариоцитарный лейкоз.
В группу хронических лейкозов включены заболевания, морфологический субстрат которых представлен созревающими и зрелыми гемопоэтическими клетками (см. схему кроветворения). В зависимости от такой опухолевой дифференцировки классификация хронических лейкозов включает:
1) хронический миелолейкоз;
2) сублейкемический миелоз (первичный миелофиброз);
3) хронический моноцитарный лейкоз;
4) хронический миеломоноцитарный лейкоз;
5) хронический мегакариоцитарный лейкоз;
6) истинная полицитемия (эритремия);
7) хронический лимфолейкоз;
8) парапротеинемические гемобластозы.
Клиническая классификация в этих заболеваниях выделяет доброкачественные и злокачественные опухоли. Доброкачественные формы на протяжении длительного времени характеризуются монотонным течением, злокачественные – характеризуются опухолевой прогрессией с клиническим динамизмом.
Этиология. Единой причины гемобластозов не существует. Система крови обладает высокой митотической активностью гемопоэтической ткани, высокочувствительна к действию повреждающих факторов. Развитие гемобластозов – сложный процесс, обусловленный сочетанием воздействия многообразных внешних и внутренних факторов. К ним относятся: ионизирующее излучение (ИИ); токсические воздействия химических веществ; вирусы; хромосомные аномалии. Связь возникновения лейкозов с ИИ обнаружена в Японии у людей, подвергшихся атомной бомбардировке; ВОЗ обозначает радиационно-индуцированный лейкоз у лиц с дозовой нагрузкой свыше 1 Грея. Эпидемиологические исследования указывают на лейкозогенное действие многих химических веществ – бензола, ароматических аминов, полициклических ароматических углеводородов и т.д., а также и лекарственных препаратов, включая ряд цитостатиков. Получены доказательства связи с вирусами лимфомы Беркита и Т-клеточного лейкоза. Определено значение генетических факторов, о чём свидетельствует увеличение заболеваемости лейкозами при некоторых генетических нарушениях и аномалиях развития – болезни Дауна, Клайнфелтера, анемии Фанкони и т.д.
Патогенез.В норме кроветворение происходит островками, состоящими из клеток определённого вида; пролиферация и дифференцировка клеток происходит параллельно. Стволовые кроветворные клетки (СКК) закладываются онтогенетически, имеют высокий, но лимитированный пролиферативный потенциал и обладают способностью:
а) самоподдержания,
б) пролиферации,
в) дифференцировке по всем росткам (см. рис.1 «Схему кроветворения»).
Считается, что у человека около 0,3-0,7% клеток КМ относятся к стволовому пулу. Ежедневно продуцируется: 24х1010 — эритроцитов, 12х1010 — гранулоцитов, 15х1010 — тромбоцитов, 2х1010 — лимфоцитов. В гемопоэтической ткани доказано возникновение опухолевого процессаиз одной мутировавшей клетки, ее потомство (клон) метастатическим путем поражает органы кроветворения и вызывает проявления заболевания.
Единичного генетического повреждения клетки костного мозга недостаточно для превращения её в опухолевую. Накопление ряда мутаций в течение продолжительного времени, часто многих лет, приводит к злокачественному новообразованию. Генетические нарушения способствуют либо активации протоонкогенов, стимулирующих пролиферацию клеток, либо подавление генов – супрессоров опухолевого роста, тормозящих пролиферацию. Опухолевые клетки теряют ферментативную специфичность, становятся морфологически и цитохимически недифференцируемыми и неправильная работа генов контролирующих рост, деление клеток вызывают неконтролируемую клеточную пролиферацию. Происходит смена дифференцированных клеток бластными (патологическими), что ведёт к качественному изменению кроветворения, в котором недифференцированные бластные клетки угнетают нормальные ростки. Развиваются анимический, геморрагический и/или иммунодефицитный синдромы.
| Рис. 1. Схема кроветворения |
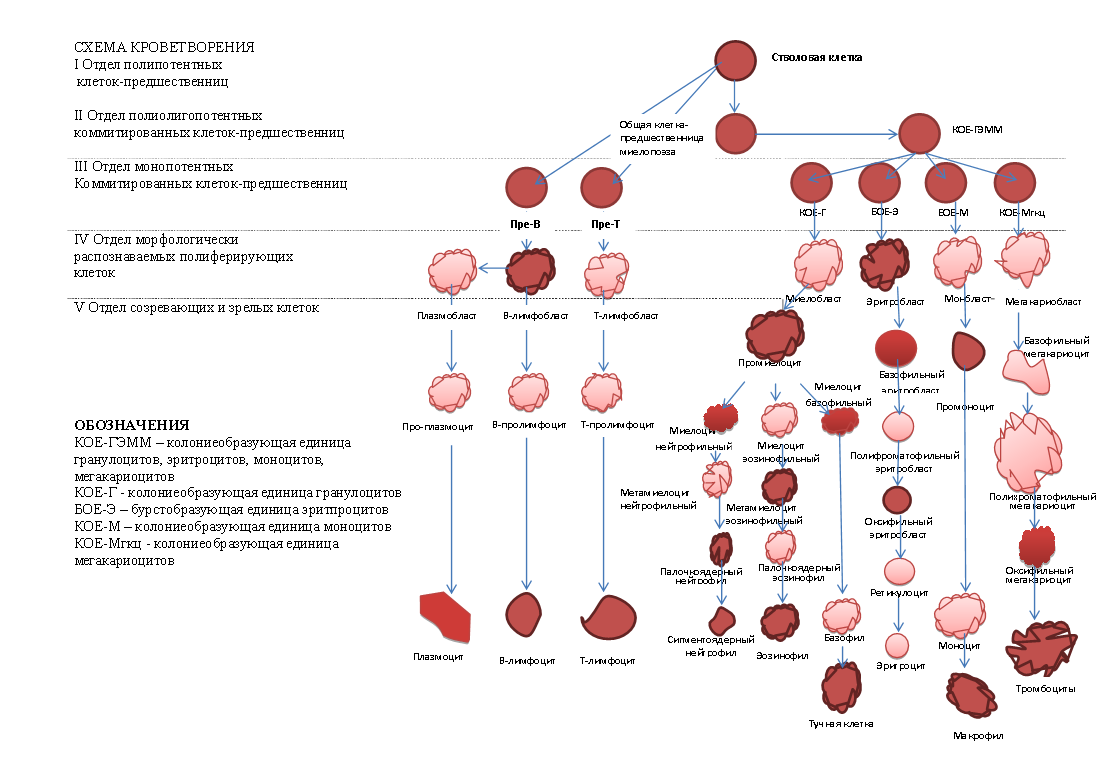 По мере прогрессирования заболевания нарастает масса опухолевых клеток с клиническими проявлениями лимфаденопатии. В дальнейшем в основном клоне гемобластоза, например хронического миелолейкоза, закономерно образуются новые опухолевые субклоны клеток с вовлечением в патологический процесс III и/или II уровня гемопоэза, отмечается переход в т.н. поликлональную стадию. Появление опухолевых субклонов отражается экстрамедуллярными метастазами, развитием резистентности к ранее эффективному цитостатическому лечению и означает качественно новый этап развития заболевания.
По мере прогрессирования заболевания нарастает масса опухолевых клеток с клиническими проявлениями лимфаденопатии. В дальнейшем в основном клоне гемобластоза, например хронического миелолейкоза, закономерно образуются новые опухолевые субклоны клеток с вовлечением в патологический процесс III и/или II уровня гемопоэза, отмечается переход в т.н. поликлональную стадию. Появление опухолевых субклонов отражается экстрамедуллярными метастазами, развитием резистентности к ранее эффективному цитостатическому лечению и означает качественно новый этап развития заболевания.
Лейкозы и лимфомы объединяет гистогенетическое родство исходных кроветворных клеток и взаимный переход: лейкозы могут сопровождаться опухолевыми разрастаниями вне костного мозга - саркоматизация лейкоза, а прогрессирование лимфомы и ее диссеминация приводят к поражению костного мозга - лейкемизация лимфом.
В гемобластозах патогенетическую особенность имеют опухоли, развивающиеся из иммунокомпетентных клеток (лимфоцитов и плазмоцитов), синтезирующих иммуноглобулины, ответственных за гуморальный иммунитет. Возникший из одной мутировавшей кровяной клетки В-лимфоцитарного ряда клон опухолевых клеток постепенно увеличивается и продуцирует в возрастающем количестве патологический иммуноглобулин. Парапротеин не обеспечивает полноценную иммунную защиту организма, ведет к синдрому белковой патологии, повышенной вязкости, висцеральным поражениям. К таким парапротеинемическим опухолям относят множественную миелому, макроглобулинемию Вальденстрема и болезнь тяжелых цепей.
Острые лейкозы (ОЛ) – первично поражающие костный мозг опухоли системы крови, субстратом которых являются бластные клетки.
ОЛ составляют 3% всех опухолевых заболеваний. В РБ заболеваемость ОЛ составляет 8 на 100 тыс. взрослого населения (18 лет и старше).
Классификация. Существуют различные классификации ОЛ. В связи с особенностью клинического течения, выбора тактики лечения ОЛ разделяют на группы лимфобластных и нелимфобластных (миелоидных) лейкозов. По морфологической и цитохимической характеристике бластов в группе острых лимфобластных лейкозов (ОЛЛ) выделено 3 формы: L1, L2, L3. В соответствии с классификацией ВОЗ 2001г. выделяются два варианта ОЛЛ:
1) острый В-лимфобластный лейкоз (лимфома из клеток-предшественниц);
2) острый Т-лимфобластный лейкоз (лимфома из клеток-предшественниц).
Критерием разделения ОЛЛ и лимфобластной лимфомы является бластоз костного мозга:
< 25% - лимфома, > 25% -ОЛЛ, хотя лечение идентично. В острых миелобластных лейкозах (см. классификацию гемобластозов) выделяют 8 вариантов: Мо, М1-М7, в которых Мо – острый недифференцируемый лейкоз, который происходит из полипотентной клетки-предшественницы, но с терапией по протоколам для миелобластных лейкозов. Важную роль имеет иммунологическая характеристика бластных клеток определяемая по набору их поверхностных дифференцировочных антигенов – CD (сlusterofdifferentiation). Такая характеристика указывает на тип патологической клоновой клетки, стадии дифференцировки, на котором произошло развитие лейкоза, что влияет на прогноз заболевания и подбор дифференцированного лечения.
Для определения тактики лечения и прогноза заболевания важное клиническое значение имеет установление стадии ОЛ. Выделяются следующие стадии заболевания.
I. Начальная стадия. Отмечаются стойкие изменения в крови: анемия, лейкоцитоз или лейкопения с нейтропенией, клиническая симптоматика отсутствует и диагностируется обычно ретроспективно.
II. Развёрнутая стадия. Характеризуется выраженным угнетением нормального кроветворения и высоким бластозом костного мозга с клинической картиной, характерной для ОЛ.
III. Ремиссия. Полное отсутствие клинической симптоматики. Нормализация гематологических показателей периферии крови (отсутствие бластных клеток, гранулоцитов > 0,5. 109/л, тромбоцитов >75.109/л, Нв >110 г/л) и костного мозга (бластных клеток менее 5%, восстановление 3-х ростков гемопоэза).
IV. Выздоровление. Состояние полной клинико-гематологической ремиссии и отсутствие признаков болезни на протяжении 5 и более лет.
V. Рецидив. Выход остаточной лейкозной популяции лейкозных клеток из-под контролирующего действия терапии. Может быть костномозговым с определением более 5% бластов в пунктате, или местным с появлением экстрамедуллярного лейкемического очага.
Клиническая картина.Вначальном периоде заболевание протекает бессимптомно с сохраненной работоспособностью. Появляются стойкие изменения в крови: анемия, лейкоцитоз или лейкопения с нейтропенией. В этом случае ОЛ может выявляться по анализам крови, проводимым в связи с профилактическим осмотром или обследованием по поводу другого заболевания. Спустя несколько недель с постепенно развивающейся общей слабостью, недомоганием, субфебрильной температурой, болями в костях и суставах, т.е. неспецифическим интоксикационным синдромом заболевание переходит в развернутую стадию. ОЛ в развёрнутой стадии характеризуют 4 основные синдрома.
Интоксикационный синдром (повышение температуры тела, общая слабость, утомляемость, потливость, снижение массы тела) обусловлен лизисом бластных клеток.
Анемический синдром (бледность кожи и слизистых, повышенная утомляемость, головокружение, систолический шум, тахикардия, слабость и др.) связан с угнетением нормального кроветворении при лейкозной гиперплазии и инфильтрации бластами костного мозга.
Гиперпластический синдром: региональная или генерализованная лимфаденопатия, при которой ЛУ плотные, безболезненные при пальпации, не спаяны межу собой и с окружающими тканями; гепато-, спленомегалия, что характерно для ОЛЛ; гиперплазия миндалин, десен и язвенно-некротические изменения в полости рта, что характерно для ОМЛ; лейкемиды – инфильтраты в дерме в виде красновато-синеватых папулообразных бляшек; поражение костной ткани (оссалгии) и др. При ОЛ такие внекостномозговые поражения обусловлены очагами лейкемической пролиферации. Особое значение имеет поражение нервной системы. Возникновение нейролейкемии обусловлено метастазированием лейкозных клеток в оболочки головного и спинного мозга или в вещество мозга. В неврологическом статусе возможны проявления различной тяжести — от лёгкой общемозговой симптоматики (головная боль, головокружение) до тяжелых очаговых поражений (нарушение сознания, снижение остроты зрения, дисфазия, менингиальный синдром. Выявляется исследованием спинно-мозговой жидкости:при нейролейкемии в ликворе определяется высокий цитоз за счет бластных клеток.
Геморрагический синдром (петехиально-пятнистый тип кровоточивости с подкожными, подслизистыми кровоизлияниями, десневыми, носовыми, маточными кровотечениями) обусловлен угнетением мегакариоцитарного ростка с тромбоцитопенией и нарушением первичного гемостаза.
Диагноз и дифференциальный диагноз. Анамнестические данные и клинические синдромы позволяют заподозрить ОЛ. Лабораторная диагностика устанавливает диагноз, но только после выявления в крови и костном мозге пациентов бластных клеток. Гематологические показатели ОЛ:а) рано выявляется нормо- или гипохромная анемия легкой или средней степени тяжести;б) при лейкемических формах острого лейкоза отмечается высокое количество лейкоцитов – до 100х109/л. и более; при сублейкемических формах - лейкоцитов 20-25х109/л, а при алейкемических – в пределах нормы или выше;в) в типичных случаях расширение лейкоцитов за счёт юных форм, наличие бластов сотсутствием промежуточных форм нейтрофилов (промиелоцитов, миелоцитов, метамиелоцитов), разрыв которых носит название «лейкемического зияния».
Во всех случаях ОЛ при исследовании пунктата костного мозга выявляется большой процент бластных клеток – от 25-30% до полного замещения нормального кроветворения. В специализированных отделениях с помощью цитохимических, цитогенетических исследований и иммунофенотипирования верифицируется вариант острого лейкоза.
Цитохимические реакции бластных клеток выявляют ОЛЛ и ОМЛ лейкозы. При ОЛЛ отмечается положительная ШИК-реакция на наличие гликогена и реакция на кислую фосфатазу; при остром В-клеточном лимфобластном лейкозе Шифф-йодная кислота в большинстве клеток выявляет гранулы гликогена вишнево-фиолетовым окрашиванием; Т-клеточные ОЛЛ реакция на кислую фосфатазу положительная в виде крупных красных гранул. При ОМЛ маркером бластов гранулоцитарного ряда являются реакции на пероксидазу, с окрашиванием гранул в желтый цвет, и реакции на липиды с использованием судана В, которые окрашивают их в черный цвет. Высокая активность неспецифических эстераз, ингибируемая фторидом натрия, используется для идентификации моноцитарного ОЛ.
Иммунологические методы идентификации бластных клеток показали вариабельность цитохимических реакций ОЛ. В целях выявления типа острого лейкоза широко используют такие методы как Е-РОК, прямая иммунофлюоресценция, выявляющая поверхностные (Smlg) и внутриклеточные (Clg) иммуноглобулины, моноклональные антитела к различным антигенным детерминантам клеток. Иммунофенотипирование в диагностике форм ОЛ занимает важное место. На поверхности гемопоэтических клеток определено около 350 антигенов, названных кластерами дифференцировки. С помощью моноклональных AT определяя наличие или отсутствие CD-маркёров лимфоидных и миелоидных и др. антигенов проводят точную диагностику ОЛЛ, а в случаях морфологически недифференцируемых лимфобластных и миелобластных лейкозов их дифференциальную диагностику. Иммунофенотипическое исследование дополняет стандартную морфоцитохимическую диагностику и позволяет уточнять варианты ОЛЛ и ОМЛ.
В ряде случаев используется электронная микроскопия, например для идентификации микрогранулярного промиелоцитарного лейкоза - подтипа ОМ3.
Острый лейкоз необходимо дифференцировать с хроническими лейкозами, злокачественными лимфомами в стадии лейкемизации, апластической анемией, метастазами рака в костный мозг, инфекционным мононуклеозом.
Примеры формулировки диагноза:
1. Острый лимфобластный лейкоз, L2 В-клеточный вариант, атака первая, индукция ремиссии, геморрагический синдром.
2. Острый миелобластный лейкоз с созреванием (ОМЛ-М2), консолидация ремиссии,клинико-гематологическая субкомпенсация.
Лечение. Пациенты с впервые выявленным острым лейкозом госпитализируются в специализированный стационар. При необходимости в терапевтическом отделении проводится только симптоматическая терапия (трансфузии эритроцитарной массы, дезинтоксикационные средства, антибиотики). В условиях гематологического отделения проводится верификация диагноза с установлением формы заболевания и его лечение по специальной программе. Лечение острых лейкозов включает:
1) химиотерапию;
2) симптоматические средства;
3) трансплантацию костного мозга.
В химиотерапии ОЛ различают:
1) индукцию (достижение) ремиссии,
2) консолидацию (закрепление) ремиссии,
3) поддерживающую терапию. Максимально полное удаление (эрадикация) бластных клеток достигается циклическим применением программ лечения. В программы включают ряд цитостатических препаратов с учетом характера их действия на различные фазы клеточного цикла патологических клеток: рубомицин (доксорубицин), циклофосфан, викристин, цитозар, метотрексат, вепезид, 6-меркаптопурин (тиогуанин), L-аспарагиназа согласно унифицированным протоколам. Например, в лечении ОМЛ основными препаратами являются цитозар и рубомицин по программе «7+3» - цитозар по 100 мг/м2/сут инфузионно в течение 7 дней и в течение 3 дней рубомицин по 45 мг/м2/сут. Во время индукции ремиссии ОЛ проводится обычно не менее двух курсов полихимиотерапии. На период индукции ремиссии, в целях профилактики инфекционных осложнений, пациента помещают в изолятор или стерильный бокс, проводят санацию ЖКТ, ежедневную обработку кожи и слизистых, профилактику геморрагических, неврологических и др. осложнений. В период консолидации используются схемы цитостатической терапии, приведшей к достижению ремиссии. Поддерживающая терапия включает проведение раз в 1-2 месяца очередного курса химиотерапии. Комплексная терапия острых лейкозов преследует цель - добиться полной ремиссии, которая должна подтвердиться по 4-м критериям:
1) в периферии крови — отсутствуют бласты, гемоглобин >100 г/л, гранулоциты >1500, тромбоциты >100х109/л;
2) в костном мозге — бластных клеток ≤5%;
3) в клинике — нет болезненных симптомов;
4) субъективно — отсутствуют жалобы.
Несмотря на поддерживающую терапию, имеется риск развития рецидива из-за неполной эрадикации клеток лейкозного клона во время индукции ремиссии, их последующей пролиферации. В целях повышения эффективности лечения ОЛ все большее распространение находит трансплантация костного мозга, полученного от HLA-совместимых доноров (аллогенный костный мозг). Предтрансплантационная подготовка включает введение сверхбольших доз цитостатических препаратов, в ряде случаев сочетающееся с тотальным облучением тела в дозе 8-10 Гр. Миелотрансплантация обычно проводится во время первой ремиссии.
Хронический лимфолейкоз (ХЛЛ) - опухоль системы крови, возникающая из костно-мозговой клетки – предшественницы лимфопоэза. Основным субстратом опухоли являются зрелые лимфоциты.
Особенностью хронического лимфолейкоза являются частые вторичные иммунодефицитные состояния и аутоиммунные осложнения. Заболевание развивается у лиц старше 50-60 лет.
Этиология и патогенез. Заболевание развивается по общим с другими гемобластозами этиологическими факторами и патогенетическим механизмам, вместе с тем в развитие ХЛЛ большое значение имеет наследственная предрасположенность – заболевание возникает у кровных родственников в нескольких поколениях. Как и все лейкозы, ХЛЛ является клональной опухолью. В опухолевой процесс могут вовлекаться различные субпопуляции лимфоцитов, что обуславливает многообразие вариантов заболевания и подтверждается определением хромосомных маркеров. Иммунофенотипирование лейкозных клеток выделяет В-формы (95% всех случаев хронического лейкоза) и Т-формы (5%). В патогенезе заболевания большое значение имеют нарушения иммунологического статуса: снижение уровня Т-супрессоров, натуральных киллеров, гипогаммаглобулинемия, развитие аутоиммунных цитопений.
Классификация. Выделяют 3 стадии хронического лейкоза:
1) начальная – умеренное увеличение периферических лимфатических узлов, лейкоцитоз не выше 30-50х109/л;
2) развернутая – прогрессирующий рост лимфатических узлов, печени, селезенки, нарастающий лейкоцитоз, развиваются инфекционные осложнения;
3) терминальная – злокачественная трансформация заболевания.
Клиника. Начальная стадия характеризуется вариабельностью течения. В ряде случаев длительно, в течение многих лет состояние не нарушается, в периферической крови отмечается умеренный лимфоцитоз в сочетании с несколько повышенным уровнем лейкоцитов. Возможно прогрессирующее течение, при котором лейкоцитоз и размеры лимфоидных органов увеличиваются быстро, рано развиваются осложнения. При доброкачественном варианте хронический лимфолейкоз прогрессирует медленно в течение нескольких лет с постепенным ростом размеров лимфатических узлов, селезенки, печени. При исследованиях периферии крови отмечается постепенное увеличение лейкоцитов (более 50-100х109/л) и лимфоцитов (80-90%) с характерными полуразрушенными ядрами лимфоцитов (тени Гумпрехта-Бехтерева). Появляются признаки опухолевой интоксикации: утомляемость, общая слабость, потливость. Для ХЛЛ характерны инфекционные осложнения бактериального (пневмонии, нагноения, сепсис) и вирусного (герпетического) генеза. Они развиваются из-за нарушений гуморального и клеточного иммунитета и являются основной причиной летальных исходов. Другим вариантом осложнений заболевания являются цитопении, которые развиваются по ряду причин: - тотальная лимфоцитарная инфильтрация костного мозга, гиперспленизм, аутоантитела к эритроцитам и (или) тромбоцитам. При ХЛЛ возможна злокачественная трансформация с переходом в неходжкинскую лимфому высокой степени злокачественности, или развитие второй злокачественной гематологической (острый лейкоз) или негематологической (рак) опухоли.
Диагноз заболевания считается достоверным при стойком абсолютном лимфоцитозе периферической крови (выше 10х109/л) и костного мозга (более 30%) с симптоматикой увеличения периферических лимфатических узлов. Дифференциальный диагноз проводится с лимфомами – лимфогрануломатозом (лимфома Ходжкина) и злокачественными неходжкинскими. Лимфаденопатия при обеих форм лимфом сочетается с повышенной температурой (характерна лихорадка). В крови: при лимфоме Ходжкина - характерно развитие нейтрофильного лейкоцитоза, лимфопении, резкое повышение СОЭ; при неходжкинской лимфоме – умеренный лейкоцитоз, уменьшение СОЭ. Окончательный ответ дают результаты гистологического исследования лимфатического узла, но его недостаточно для установления диагноза ХЛЛ, поскольку аналогичная картина крови и костного мозга может наблюдаться при некоторых видах лимфом. Современные критерии диагноза ХЛЛ включают также иммунологические исследования: лимфоциты при ХЛЛ имеют абсолютно характерный иммунофенотип - антигены CD19, CD5, CD23 и антигены CD20 и CD22.
Примеры формулировки диагноза:
1. Хронический лимфолейкоз, начальная стадия.
2. Хронический лимфолейкоз, развернутая стадия, клинико-медикаментозная компенсация.
Лечение. В начальной стадии пациенты с хроническим лимфолейкозом подлежат наблюдению, контролируя гемограмму 1 раз в 6 месяцев. Важное значение придается соблюдению режима труда и отдыха, витаминотерапии, исключаются гиперинсоляции и переохлаждения. В ряде случаев прогрессирование заболевания отсутствует в течение многих лет. Показаниями к началу лечения служат: ухудшение общего состояния с быстрым увеличением лимфатических узлов, селезенки, печени, или синдрома компрессии; нарастающий лейкоцитоз более 150х109/л. или удвоение абсолютного числа лимфоцитов в крови менее чем за 12 месяцев; развитие аутоиммунной анемии или тромбоцитопении, наличие комплексных хромосомных аберраций. Лечение направлено на снижение опухолевой массы, количества лейкоцитов в периферической крови, уменьшение явлений интоксикации. Основным методом лечения прогрессирующего ХЛЛ является химиотерапия. Выбор химиопрепарата: хлорбутин (лейкеран) - при высоком лейкоцитозе, циклофосфан - при преимущественном увеличении лимфатических узлов, или флударабин осуществляет гематолог. Новым и важнейшим этапом в лечении ХЛЛ стало появление и внедрение в клиническую практику моноклональных антител. Первым в терапии ХЛЛ стал применяться препарат ритуксимаб (Мабтера) — моноклональные антитела к антигену CD20. Ритуксимаб (375 мг/м2 или 500 мг/м2)эффективен в монотерапии в больших дозах, а также в сочетании ритуксимаба и флударабина - частота ремиссий у ранее леченных составляет 60–70%, у нелеченых — 90–95%.
Хронический миелолейкоз – опухоль системы крови, возникающая из клетки – предшественницы гемопоэза, которая сохраняет способность дифференцироваться до зрелых форм. Морфологический субстрат опухоли составляют преимущественно нейтрофильные гранулоциты.
Заболевание может возникать в любом возрасте, но чаще у лиц 30 – 70 летнего возраста.
Этиология и патогенез хронического миелолейкоза соответствуют общим закономерностям возникновения и развития гемобластозов. Одним из доказательств клонового характера заболевания является обнаружение в клетках костного мозга более чем у 95% пациентов специфической хромосомной аномалии – хромосома из 22 пары с укороченным длинным плечом. Эта аномалия (филадельфийская, Ph-хромосома) возникает вследствие реципрокной транслокации между 9-й и 22-й хромосомами. Патологический клон (опухолевой) медленно увеличивается и длительно не проявляется симптоматикой. По мере прогрессирования заболевания и нарастания массы опухолевых клеток появляются общие симптомы и клинико-гематологические проявления. В дальнейшем в основном клоне хронического миелолейкоза закономерно образуются новые опухолевые субклоны клеток II и/или III уровня гемопоэза. Возникшие новые клеточные клоны более злокачественны, значительно хуже поддаются лечению и приводит к развитию терминальной стадии лейкоза.
Классификация. Выделяют три стадии хронического миелолейкоза:
1) начальная – миелоидная пролиферация костного мозга с изменениями в периферической крови без явлений интоксикации;
2) развернутая – выраженная миелоидная пролиферация костного мозга с изменениями в периферии крови и клиническими проявлениями с интоксикацией, спленомегалией;
3) терминальная – рефрактерность к проводимой цитостатической терапии, значительное увеличение селезенки, развитие бластного криза или экстрамедуллярных саркоматозных очагов.
Клиника. В начальной стадии клинических проявлений нет. В периферической крови обнаруживается умеренный нейтрофильный лейкоцитоз (обычно 12-15х109/л) со сдвигом до миелоцитов и промиелоцитов. В развернутой стадии появляются клинические признаки опухолевой интоксикации – быстрая утомляемость, потливость, в ряде случаев субфебрильная температура тела, снижение массы тела. Постоянным признаком является увеличение селезенки, иногда до значительных размеров. Отмечается гепатомегалия. Нарастает нейтрофильный лейкоцитоз. В лейкоцитарной формуле, кроме омоложения состава гранулоцитов, часто увеличен процент базофилов и (или) эозинофилов – «эозинофильно-базофильная ассоциация». Количество тромбоцитов чаще в норме, но у 20-30% пациентов с начала заболевания отмечается гипертромбоцитоз до 1,5х1010/л и более. Более чем у 90% пациентов при цитогенетическом исследовании обнаруживается Рh-хромосома. При прогрессировании заболевания, обычно через 3-4 года доброкачественная моноклоновая опухоль превращается в поликлоновую с развитием терминальной стадии с характеристикой бластного криза. В периферии крови появляется множество бластных клеток (30-90%), нарастает тромбоцитопения с развитием тяжелого геморрагического синдрома, возникают инфекционные осложнения. В терминальной стадии могут на фоне резистентности к цитостатической терапии появляться очаги саркомного роста в лимфатических узлах, коже и др. органах, равивается тяжелая оссалгия, упорная лихорадка, истощение.
Диагноз основывается на характерных данных анализа крови, результатах исследования костного мозга, увеличении размеров селезенки. Диагностические критерии хронического лейкоза:
а) лейкоцитоз более 20х109/л с расширением лейкоцитарной формулы до единичных бластных клеток с наличием всех созревающих форм нейтрофильного ряда, а также эозинофильно-базофильной ассоциации;
б) в миелограмме отмечается миелоидная пролиферация костного мозга, соотношение лейко/эритро достигает 10:1 – 20:1; в трепанате отмечается вытеснение жирового костного мозга кроветворными клетками;
в) обнаружение Рh-хромосомы;
г) спленомегалия. В развернутой стадии заболевания основным лабораторным признаком фазы акселерации является прогрессирующее увеличение бластов (от 10 до 19%) и базофилов (>20%) в крови и костном мозге. В терминальной фазе, протекающей в форме бластного криза, в крови или костном мозге определяется ≥ 20 % бластных клеток.
Дифференциальный диагноз проводится с лейкемоидными реакциями миелоидного типа. Термин «лейкемоидная реакция» определяет изменения крови, которые напоминают картину лейкоза и могут возникать при различных инфекциях, злокачественных новообразованиях, диффузных заболеваниях соединительной ткани. Основные дифференциально-диагностические признаки лейкемойдной реакции включают: а) клинические характеристики, которые соответствуют основному заболеванию; б) эволюция изменений в крови, с повышенным количеством базофилов при хроническом лейкозе и нормальным при лейкемойдной реакции, в) Рh-хромосома отсутствует.
Примеры формулировки диагноза:
- Хронический миелолейкоз, Ph+, начальная стадия.
- Хронический миелолейкоз, Ph+, развернутая стадия (фаза акселерации), спленомегалия.
Лечение. Основные методы лечения хронического миелолейкоза являются:1) химиотерапия;2) альфа-интерферон;3) трансплантация костного мозга. Эффективно в развернутой стадии монотерапия алкилирующим средством - бусульфан по 2-4 мг/сут., в ряде случаев гидроксикарбамид по 1000- 2000 мг/сут. С прогрессированием и развитием бластного криза лечение проводится в соответствии с принципами лечения острых лейкозов. Цитостатическая терапия в лечении в развернутой стадии хронического миелолейкоза эффективна, но не предотвращает развитие терминальной стадии. Разработаны альтернативные методы – интерферонотерапия и трансплантация костного мозга. Использование альфа-интерферона показало эффективность у 70-80% пациентов в развернутой стадии, при этом у 45-50% пациентов достигается полная клинико-гематологическая, а у 10-15% - и цитогенетическая ремиссия с исчезновением из костного мозга Рh-аномального клона клеток.
Истинная полицитемия (эритремия) – хронический лейкоз, возникающий из клетки-предшественницы миелопоэза, сохранившей способность дифференцироваться по нескольким росткам кроветворения – эритроидному, гранулоцитарному, мегакариоцитарному, но преимущественно по эритроидному.
Заболеваемость полицитемией составляет 0,6-0,8 на 100 тыс. населения. Наиболее часто заболевают люди в возрасте старше 50- 60 лет.
Этиология и патогенез. Этиология неизвестна. В основе заболевания лежит клоновая природа миелопролиферации с генетическими дефектами хромосом: анэуплоидией, псевдоплоидией и структурными аберрациями. У 90% пациентов выявляется мутация гена JAK-2. Патогенез обусловлен усиленным эритропоэзом, эритроцитозом в периферической крови с развитием вторичных реологических и коагуляционных нарушений, миелоидной метаплазией в селезенке и печени, финальным истощением кроветворения с аплазией и миелофиброзом.
Классификация. Единой классификации заболевания нет. Выделяют три стадии истинной полицитемии: I - начальная; IIА – полицитемическая и IIБ- полицитемическая с миелоидной метаплазией селезенки; III- анемическая.
Клиника. Для заболевания характерно постепенное развитие. Пациенты отмечают общую слабость, покраснение кожных покровов с синюшным оттенком (плетора). У многих первым симптомом является кожный зуд, затем развивается эритромелалгия – острые жгучие боли в кончиках пальцев, купирующиеся приемом аспирина. Сосудистые и висцеральные осложнения развиваются редко. Селезёнка несколько увеличена, но пальпировать её обычно не удаётся (увеличение селезёнки обусловлено повышенной секвестрацией в ней тромбоцитов и эритроцитов). Начальная стадия характеризуется умеренным эритроцитозом в крови, панмиелозом в красном костном мозге. Продолжительность первой стадии может превышать 5 лет. Вторая развернутая (пролиферативная)стадия характеризуется выраженной плеторой, гепатоспленомегалией за счет миелоидной метаплазии этих органов, возможны тромбозы артериальных и венозных сосудов, геморрагические осложнения. В крови обнаруживают нарастающий эритроцитоз, отмечаются тромбоцитоз и нейтрофилёз со сдвигом лейкоцитарной формулы влево, увеличение содержания базофилов. В трепанобиоптате - панмиелоз с выраженным мегакариоцитозом, возможен очаговый коллагеновый миелофиброз. По мере прогрессирования (стадия IIБ) нарастают сплено- и гепатомегалия, чаще возникают тромботические и геморрагические осложнения. В крови - панцитоз со сдвигом лейкоцитарной формулы до миелоцитов. Продолжается вторая стадия до 15 лет. Третья анемическая стадия является исходом заболевания. Выражено увеличены печень и селезёнка, в них обнаруживают миелоидную метаплазию. В крови нарастает анемия, панцитопения. В красном костном мозге распространенный миелофиброз с сохраненным или редуцированным миелопоэзом. Кроме того, исходом полицитемии могут быть острый лейкоз, хронический миелолейкоз, апластическая анемия.
Диагноз. Клинический анализ крови с повышенным уровнем гематокрита, гемоглобина и эритроцитов. В миелограмме в пролиферативной стадии — гиперплазия трёх ростков кроветворения, патологические мегакариоциты, отсутствие отложений железа (более чем в 95% случаев), признаки начинающегося фиброза (повышенное содержание ретикулина). Однако в критерии диагноза выделен ряд признаков по категориям А и В.
Категория А:
1) увеличение массы циркулирующих эритроцитов;
2) нормальное насыщение артериальной крови кислородом (более 92%);
3) увеличение селезёнки.
Категория В:
1) лейкоцитоз более 12х109/л (при отсутствии температурной реакции, инфекций);
2) тромбоцитоз более 400х109/л.;
3) увеличение содержания щелочной фосфатазы нейтрофилов;
4) увеличение ненасыщенной витамин В12-связывающей способности сыворотки крови.
Диагноз истинной полицитемии достоверен при наличии трёх признаков категории A, или двух признаков категории А и одного признака категории В.
Дифференциальный диагноз проводится с вторичными симптоматическими эритроцитозами - относительными и абсолютными. Относительные эритроцитозы возникают с повышенной потерей жидкости при длительной рвоте, ожогах; отмечается уменьшение объема плазмы и относительное преобладание эритроцитов в единице объема крови, а масса циркулирующих эритроцитов не изменена. Абсолютные эритроцитозы связаны с повышением образования эритропоэтинов, характеризуются повышенным эритропоэзом и увеличением массы циркулирующих эритроцитов. Встречаются при гипоксических состояниях (при заболеваниях легких, врожденных пороках сердца), некоторых опухолях (гипернефрома, опухоли надпочечника, гепатома) и заболеваниях почек (поликистоз, гидронефроз, стеноз почечных артерий).
Примеры формулировки диагноза:
1. Истинная полицитемия, I стадия.
2. Истинная полицитемия, ПБ стадия. Гепатоспленомегалия. Эритромелалгии. Микроциркуляторная энцефалопатия.
3. Истинная полицитемия, III стадия. Анемический синдром. Острый тромбофлебит вен правой голени. Гепатоспленомегалия. Прогрессирующая кахексия.
Лечение. Для лечения полицитемии используют два основных метода:
а) кровопускания (гемоэксфузии),
б) химиотерапия с активной симптоматической терапией.
Кровопускание применяют при плеторическом синдроме для уменьшения массы циркулирующих эритроцитов. Удаляют по 500 мл крови через 1-2 дня (у лиц пожилого возраста и при наличии сопутствующих заболеваний удаляют не более 350 мл крови). Непосредственно перед кровопусканием вводят 400 мл реополиглюкина в/в капельно и 5000 ЕД гепарина. Процедуры проводят до снижения концентрации НЬ до 140—150 г/л, гематокрита — до 46—47%. При этом значительно снижается риск сосудистых осложнений, уменьшается кожный зуд. Вместо кровопускания можно проводить эритроцитаферез. Показания к назначению цитостатической терапии являются панцитоз, спленомегалия, висцеральные, сосудистые осложнения. Препарат выбора — гидроксикарбамид. По сравнению с другими цитостатиками он оказывает менее выраженное мутагенное действие и вызывает меньше побочных эффектов. Препарат назначают по 40—50 мг/кг/сут в 2—3 приёма (по 2—3 капсулы в день). Во время лечения необходим контроль содержания лейкоцитов. Гидроксикарбамид сочетают с альфа-интерфероном в дозе 3—5 млн ME п/к 3—7 раз в неделю длительно (не менее года).
Антиагрегантную терапию проводят для профилактики тромбозов. Используют ацетилсалициловую кислоту по 300-500 мг/сут или дипиридамол по 150— 200 мг/сут. Во время лечения необходим контроль за агрегацией тромбоцитов. Для уменьшения уратового диатеза назначают аллопуринол. При развитии аутоиммунной анемии или тромбоцитопении назначаются глюкокортикоиды.
Лимфогрануломатоз (Лимфома Ходжкина) – первично опухолевое заболевание лимфатической системы, распространяющееся метастатическим путем.
Показатель заболеваемости лимфомы Ходжкина (ЛХ) в мире в последние десятилетия увеличился до 4. В Республике Беларусь лимфома Ходжкина в структуре онкологической заболеваемости составляет 0,8-0,9%. Стандартизованные показатели заболеваемости составили 2,7 на 100 000 населения. Заболевание встречается в любом возрасте, однако существует два пика заболеваемости – в 20-29 лет и после 60 лет. Лица мужского пола составляют 60-70%, преимущественно за счет молодого возрастного периода.
Этиология и патогенез. Этиологический фактор не установлен. Ряд авторов этиологию ассоциируют с вирусом Эпштейна-Барра, т.к. в 20% клеток Березовского-Штернберга находят генетический материал этого вируса. В возникновении заболевания существуют вирусная, генетическая и иммунологическая теории. Источник опухоли ЛХ неизвестен. По данным иммунофенотипирования характерные для ЛХ многоядерные клетки Березовского-Штейнберга несут на своей поверхности антигены характерные как для лимфоидного ростка, так и для моноцитарного ростка (СD25, CD20, CD15 CD30 и др). В патогенезе заболевания существенную роль отводят иммунному дефекту, связанному с нарушением функции Т-лимфоцитов и нарастающему по мере прогрессирования болезни. При ЛХ процесс возникает мультицентрически одновременно в нескольких точках, которые сливаются в одно опухолевое поле, занимающее часть или весь лимфоидный орган, обычно лимфатический узел. Распространение процесса происходит лимфогенным путем с поражением смежных групп лимфатических узлов и органов. Значительно реже отмечается гематогенное метастазирование, которое может сочетаться с лимфогенной диссеминацией. В случае возникновения опухолевых очагов в нескольких лимфатических узлах или органах, заболевание проявляется на поздней стадии и характеризуется неблагоприятным течением.
Классификация. В клинике приняты клиническая и гистологическая классификации. Клиническая картина ЛХ в классификации ВОЗ учитывает местные и общие проявления и выделяет 4 стадии:
Iстадия - поражение одной лимфатической области или лимфатической структуры (I) или локальное поражение одного экстралимфатического органа или ткани (IE).
II стадия - поражение двух или более лимфатических областей по одну сторону диафрагмы (II) или локализованное поражение одного экстралимфатического органа или ткани и их регионарных лимфатических узлов с или без поражения других лимфатических узлов по ту же сторону диафрагмы (IIE).
III стадия - поражение лимфатических узлов по обе стороны диафрагмы (III), которое может сочетаться с локализованным поражением одного экстралимфатического органа или ткани (III E), или с поражением селезенки (IIIS), или с поражением того и другого (III E + S).
IV стадия - диссеминированное (многофокусное) поражение одного или нескольких экстралимфатических органов с или без поражения лимфатических узлов; или изолированное поражение экстралимфатического органа с поражением отдаленных (не регионарных) лимфатических узлов.
Каждая стадий подразделяется на 2 подстадии в зависимости от отсутствия (А) или наличия (Б) хотя бы одного из общих симптомов (лихорадка неясного генеза >38°С продолжительностью не менее 1 нед., профузные ночные поты, снижение массы тела на 10% и более за последние 6 мес.) и биохимических показателей активности.
Гистологическая классификация (ВОЗ,1999 г.) выделяет 5 морфологических вариантов. Заболевание включает два основных морфологических варианта:I) - классическая лимфома Ходжкина; II)- нодулярная ЛХ с преобладанием лимфоцитов, т.е. вариант с большим количеством лимфоцитов (встречается редко).
Классическая ЛХ включает 4 основные варианта:
1) лимфоидное преобладание (3-5%), (характеризуется увеличением числа лимфоцитов и гистиоцитов, а также незначительным количеством клеток Березовского-Штернберга);
2) лимфоидное истощение (1-2%), (относительная бедность лимфоцитами и обилие клеток Березовского-Штернберга);
3) смешано-клеточный вариант (20%), (гистологические признаки с промежуточной характеристикой между лимфоидным преобладанием и лимфоидным истощением, определяется различное количество лимфоцитов, эозинофильных гранулоцитов и плазматических клеток, умеренное количество клеток Березовского-Штернберга);
4) нодулярный склероз (75%), (характеризуется коллагеновым фиброзом, окружающего островки лимфатической ткани, присутствуют клетки Березовского- Штенберга).
Между гистологическим типом ЛХ и его клинической картиной установлена связь с характеристикой течения заболевания. Благоприятное течение отмечается при варианте с лимфоидным преобладанием и нодулярным склерозом, смешанно-клеточный тип занимает промежуточное место, а вариант с лимфоидным истощением характеризуется неблагоприятным течением и обычно соответствует IV стадии заболевания.
Клиника. Клиническая симптоматика ЛХ многообразна уже в начале заболевания и может проявляться одним или несколькими клиническими признаками – лимфаденопатия, паранеопластические симптомы, экстранодальные поражения: спленомегалия, гепатомегалия, поражение костного мозга, поражение легких, поражение кожи, поражение костной системы, поражение ЖКТ и/или ЦНС. Преимущественное поражение лимфатической системы или органа определяет клиническую картину заболевания.
Местные проявления лимфомы Ходжкина обусловлены локализацией и размерами пораженных лимфатических узлов и других патологических очагов в различных органах и тканях. В большинстве случаев это стойкое увеличение периферических лимфатических узлов (ЛУ) той или иной группы. Поражение лимфатических узлов выше диафрагмы встречается почти в 90% случаев и только у 10% больных изменения наблюдаются в поддиафрагмальных лимфатических коллекторах. Первое проявление ЛХ в 60-75% случаев в шейно-надключичных лимфатических узлах. Вначале увеличение (1,0 см. и более) периферических лимфатических узлов не сопровождается нарушением самочувствия пациента. При осмотре такие увеличенные лимфатические узлы подвижные, плотно-эластичные, не спаяны с кожей. У 15-20% больных заболевание начинается с увеличения лимфатических узлов переднего средостения, которое обнаруживается при флюорографии, или проявляется клинически. К числу сравнительно частых симптомов относятся кашель, вначале незначительный, сухой и боли в грудной клетке, одышка, в поздних стадиях симптомы сдавления верхней полой вены. Генерализация ЛХ сопровождается появлением симптомов интоксикации (проливные поты, потеря массы тела, лихорадка). В период развернутых проявлений заболевания возможно поражение всех лимфоидных и любых внутренних органов. У части пациентов заболевание начинается остро с лихорадки, ночных потов, похудания, а лимфаденопатия выявляется позднее.Течение ЛХ различно – от доброкачественного затягивающегося на десятки лет, до подострого с летальным исходом за несколько месяцев. Прогноз определяется стадией, гистологическим типом заболевания, выраженностью общих симптомов. При появлении признаков интоксикации с характеристикой подстадии Б прогноз резко ухудшается.
Диагноз лимфомы Ходжкина устанавливается на основе:
а) клинической картины (лимфаденопатия, спленомегалия, подавление иммунитета, нарушения функции органов, в которых развивается лимфома Х,или анемия и паранеопластические симптомы);
б) обнаружения клеток Березовского-Штернберга в материале биопсии ЛУ или органа, пораженного лимфомой. Специфических для ЛХ изменений периферической крови не существует.
У большинства отмечается умеренный нейтрофильный лейкоцитоз, иногда, высокая эозинофилия. Ускоренное СОЭ, увеличение уровня фибриногена, альфа-2-глобулинов (за счет церулоплазмина и гаптоглобина) – коррелируют с активностью процесса. В 10% при ЛХ поражается костный мозг, что обнаруживается при трепанобиопсии. Исследование миелограммы проводится при развитии выраженной цитопении или вторичного онкогематологического заболевания.
Дифференциальный диагноз при увеличении периферических ЛУ проводится с группами патологии — инфекционными заболеваниями, иммунными реакциями на введение вакцин или укусы насекомых, злокачественными лимфомами и др. формами гемобластозов.
При туберкулёзе периферических ЛУ в клинике выявляется: потливость, субфебрилитет, в ПК – лейкоцитоз, нейтрофильный сдвиг влево, эозинофилия; а также положительные результаты туберкулиновых проб.Болезнь кошачьих царапин — инфекционное заболевание, возникающее после укуса и царапин кошек. Протекает с образованием первичного аффекта в виде нагнаивающейся папулы с последующим развитием регионального лимфаденита.
Установление морфологического варианта лимфомы осуществляется путем открытой биопсии лимфатического узла и его гистологического исследования.Морфологическим субстратом любого очага при ЛХ является полиморфноклеточная гранулема, состоящая из лимфоидных (различной степени зрелости) и гистиоцитарных элементов, нейтрофилов, эозинофилов, плазматических клеток, фибробластов; в той или иной степени выражены явления склероза. Для ЛХ характерно наличие гигантских (диаметром >25 мкм до 80 мкм) клеток Березовского-Штернберга и их предшественников - клеток Ходжкина. В большинстве случаев рутинные гистологические методы недостаточно специфичны для дифференциальной диагностики внутри групп морфологически сходных лимфом, в связи с чем, проводится иммунофенотипическое определение типа лимфомы (Т- или В-клеточная) в дифференциальной диагностике с ЛХ, хромосомный анализ клеток лимфомы.
Примеры формулировки диагноза:
- Лимфома Ходжкина, вариант нодулярного склероза, IIА стадия.
2. Лимфома Ходжкина, смешанно-клеточный вариант, IIIБ стадия, состояние после 2-х курсов полихимиотерапии по схеме АВVD и лучевой терапии в суммарной дозе 30 Гр.Анемия.
Лечение пациентов лимфомой Ходжкина начинается с полихимиотерапии. Только у пациентов с IА стадией без факторов риска и гистологическим вариантом «лимфоидное преобладание» проводится R-облучение пораженной зоны в суммарной очаговой дозе (СОД) 30-36 Гр. К группе с благоприятным прогнозом относятся пациенты с I и II стадиями без факторов риска. Лечение ЛХ полихимиотерапией проводится по протоколам, например, ABVD - Доксорубицин 25 мг/м2 в/в. Блеомицин 10 мг/м2 в/в, Винбластин 6мг/м2 в/в, Дакарбазин 375 мг/м2 в/в,- дни 1-й и 14-й, с двумя курсами. После завершения химиотерапии проводится облучение зон исходного поражения. Суммарная очаговая доза (СОД) – 30 Гр при полной регрессии опухоли, 36 Гр – при частичной регрессии.
В других группах полихимиотерапия проводится по схемам, назначаемыми онкологами-гематологами. Для лечения первично-резистентных пациентов и с непрерывно-рецидивирующими формами заболевания применяется высокодозная химиотерапия под защитой аутотрансплантации костного мозга или стволовых клеток.
Современные схемы химиолучевого лечения позволяет получить пятилетнюю выживаемость у больных с благоприятным прогнозом 90-100%, с промежуточным прогнозом – 80%, с неблагоприятным прогнозом 50-70%.
Множественная миелома (ММ),миеломная болезнь — В-клеточная опухоль, характеризующаяся патологической пролиферацией В-клеток конечной стадии дифференцировки - плазматической клетки.
Заболевание составляет 10% от всех гематологических опухолей, ежегодная заболеваемость 3—5 на 100 тыс. человек. Частота ММ увеличивается с возрастом, чаще заболевание встречается в возрасте 60-70 лет.
Этиология и патогенез. Ионизирующее излучение увеличивает частоту заболеваемости. Для множественной миеломы характерны множественные хромосомные нарушения - моносомия 13-й хромосомы, трисомии хромосом 3, 5, 7, 9, 11, 15 и 19. Злокачественная трансформация происходит до стадии плазматической клетки в популяции пре-В клеток. При ММ клон опухолевых клеток сохраняет способность синтеза и секреции иммуноглобулинов и продуцирует повышенное количество иммуноглобулина (IgG, IgA, IgD, IgE) или легких цепей каппа (К) или лямбда (L). Синтезируемый парапротеин носит однородный (моноклоновый) характер. Наряду с выработкой миеломного белка опухоль секретирует фактор активирующий остекласты (OAF) – комбинация Il6,Il-1β и TNF-β. При опухолевой трансформации плазматическая клетка приобретает опухолевые свойства — способность прорастать костную ткань, почки, формируя клиническую картину распространенной многоочаговой опухоли, в небольшом проценте случаев она пролиферирует локально (солитарная плазмацитома). По мере опухолевой прогрессии моноклоновый характер опухоли сменяется поликлоновым: появляется ранее отсутствовавший белок Бенс-Джонса, или второй патологический иммуноглобулин, нарастает уровень парапротеинемии и парапротеинурии. Нарастает морфологический атипизм клеток и агрессивность опухоли, метастазирующей в различные органы, развивается её резистентность к ранее эффективной терапии.
Классификация. Клинико-анатомическая классификация выделяет диффузно-очаговую, диффузную и множественно-очаговую форму множественной миеломы. В зависимости от величины опухолевой массы выделяют 3стадии множественной миеломы. Объем опухоли расценивается как низкий, когда присутствуют все следующие признаки: а) уровень гемоглобина >100г/л; б) нормальный уровень кальция сыворотки; в) отсутствует остеолиз или солитарный костный очаг; г) низкий уровень М-компонента (IgG<50 г/л, IgА<30г/л); д) белок Бенс-Джонса в моче < 4 г/24 ч. Опухолевая масса высокая при наличии любого из следующих признаков: уровень Нb<85г/л; уровень кальция сыворотки выше нормы >1,2 г/л; обширное поражение скелета или крупные переломы; высокий уровень М-компонента (IgG>70 г/л, IgА>50 г/л); белок Бенс-Джонса в моче >12 г/24 ч. Между этими критериями объем опухоли расценивается как промежуточный.
Клиника. Заболевание проявляется несколькими основными синдромами. Костномозговой синдром обусловлен пролиферирующими в костном мозге миеломными клетками, которые вытесняют нормальные миелоидные элементы, а также приводят к деструкции костей. У большинства пациентов обнаруживается анемия с симптоматикой общей слабости, нередко развивается сердечная недостаточность. В периферии крови выявляется анемия, у ¾ пациентов значительное увеличение СОЭ до 70 – 80 мм/час, в последующем с прогрессированием заболевания и лечением – цитопении.
Поражение скелета. Рентгенологически более чем у 80% пациентов выявляются множественные очаговые дефекты костей свода черепа, тазовых костей, позвоночника, реже – проксимальных отделов трубчатых костей (плечо, бедро). Пролифелирующие в костном мозге миеломные клетки и секретирующие OAF приводят к деструктивным изменениям в костях с триадой Калера – боль, опухоли, переломы, что является типичной для ММ в поздних стадиях и агрессивных формах заболевания. В некоторых случаях первичным проявлением опухоли были симптомы сдавления спинного мозга - вынужденное положение лежа, боли в спине, усиливающиеся при движении, кашле, слабость, потеря чувствительности. Рентгенологические находки могут отсутствовать при диффузной форме поражения.
Поражение почек наиболее частое и грозное проявление гипер- и парапротеинемией. Находящиеся в плазме крови легкие цепи фильтруются через почки и повторно поглощаются и катаболизируются в канальцах. Повреждение клеток канальцев ведет к образованию белковых цилиндров. Развивается воспаление с последующим фиброзом и почечной недостаточностью — «миеломная почка». Наиболее повреждающим и продолжительным является действие парапротеина — депонирование амилоида, обнаруживаемое в почках, сердце, печени, селезенке, коже, мышцах, языке и желудочно-кишечном тракте. Амилоид нерастворим, устойчив к протеолизу и часто вызывает необратимое повреждение жизненно важных органов.
Иммунодефицит и с индром недостаточности антител развиваются из-за резкого снижения уровня нормальных иммуноглобулинов, проявляются частыми и тяжелыми бактериальными инфекциями, чаще дыхательных и мочевыводящих путей.
Синдром повышенной вязкости характеризуется парестезиями. Некоторые из М-протеинов нерастворимы при низкой температуре, а присутствие криоглобулинов вызывает синдром Рейно, ишемию пальцев и даже развитие гангрены.
Геморрагический синдром вызывается сочетанным нарушением тромбоцитарного и коагуляционного звеньев гемостаза и проявляется кровоточивостью слизистых, а также носовыми и десневыми кровотечениями.
Диагностика. У 80% пациентов выявляется анемия, выраженность которой зависит от стадии ММ. Характерно резкое ускорение СОЭ, достигающее 60-80 мм/час и более. Биохимический анализ крови: гипонатриемия, гиперкальциемия (вследствие нарушения экскреции и остеолизиса), повышение содержания креатинина, мочевины, ЛДГ. Повышение сывороточного белка выше 80 г/л за счет фракции глобулинов. На момент диагностики ММ протеинурия выявляется у 60-70% пациентов. Общие лабораторные данные, как и рентгенологически выявляемые очаги деструкции не являются основанием для диагноза, а лишь подтверждают его. Для постановки диагноза множественной миеломы необходимо получить морфологическое подтверждение опухолевого процесса плазмоклеточной природы и выявить продукт синтеза опухолевых клеток – парапротеин.
Критерии диагноза множественной миеломы:
а) в миелограмме определяется более 15% плазматических клеток;
б) парапротеинемия более 30 г/л, либо менее 30 г/л, но с выраженным снижением уровня нормальных иммуноглобулинов, либо протеинурия Бенс-Джонса более 50 мг/л. В определении классов и типов парапротеинов, а также уровня нормальных иммуноглобулинов, используют методы иммунохимического анализа.
Дифференциальный диагноз ММ проводится с заболеваниями, при которых отмечаются реактивные моноклональные иммуноглобулинопатии - группы злокачественных опухолей, диффузных заболеваний соединительной ткани, хронических активных гепатитах и циррозах печени. Для реактивных гаммапатий характерен более низкий, чем при ММ уровень парапротеинов и отсутствуют основные диагностические критерии ММ: значительный процент плазматических клеток в миелограмме и парапротеинемия.
Примеры формулировки диагноза:
1. Множественная миелома II стадия, несекретирующий тип, диффузно-очаговая форма. Компрессионный перелом L1–L2. Анемия,
2. Множественная миелома, диффузно- очаговая форма. IIIB стадия. IgG-тип секреции. Генерализованный остеодеструктивный синдром (компрессионный перелом IX и X грудных позвонков, очаговый лизис ребер, подвздошных костей, костей черепа). ХПН, стадия декомпенсации. Анемия.
Лечение. Терапия ММ включает:1) цитостатические препараты;2) глюкокортикоиды;3) лучевую терапию;4) мероприятия по предупреждению и лечению метаболических нарушений и инфекционных осложнений;5) лечебную физкультуру;6) ортопедическое и хирургическое лечение.
Основой цитостатической терапии являются алкилирующие препараты (мелфалан, циклофосфан, кармустин). Чаще проводится начальная терапия с комбинацией препаратов по протоколу М-2: преднизолон-40мг/м2 1-7дни; винкристин 1-4 мг/м2 1день; мелфалан 8 мг/м2 с 1-го по 7 дни; кармустин 20 мг/м2 1 день.Локальная лучевая терапия показана при ограниченных опухолевых узлах в костях, мягких тканей, при сдавлениях корешков спинного мозга. Лечение почечной недостаточности включает малобелковую диету, обильную гидротацию, гемосорбцию, гемодиализ. Развитие парапротеинемической комы является абсолютным показанием для проведения массивного плазмафереза. Всем пациентам рекомендуется лечебная физкультура и индивидуализированная максимально возможная физическая нагрузка.
Дата публикования: 2014-10-25; Прочитано: 6308 | Нарушение авторского права страницы | Мы поможем в написании вашей работы!
