 |
Главная Случайная страница Контакты | Мы поможем в написании вашей работы! | |
Хвороби імпринтинґу
|
|
Недавно було виявлено, що деякі гени, передані потомству, несуть специфічний «відбиток» статі одного з батьків. Іншими словами – деякі гени батька та матері проявляються у нащадків по-різному. Це явище дістало назву геномного імпринтинґу (залишання сліду).
Дитина одержує один набір хромосом з батьківським «позначенням» деяких генів, інший набір хромосом – з материнським «позначенням». При утворенні у нащадка статевих клітин попереднє «позначення» стирається, а гени визначаються заново, відповідно до статі даного організму. Внаслідок цього деякі гени, одержані від одного з батьків, перебувають в неактивному, або імпринтинґовому стані.
У людини вже знайдено понад 60 генів, які по-різному проявляються в чоловічому та жіночому організмові. Очікується, що геном людини може містити до 200 подібних генів.
Класичним прикладом хвороб імпринтинґу служать спадкові синдроми Прадера – Віллі та Ангельмана, основними клінічними проявами яких є розумова відсталість різного ступеня тяжкості в поєднанні з глибокими неврологічними розладами.
Найчастішою причиною виникнення цих захворювань є мікроделеція критичного регіону хромосоми 15 (15q11-q13). Така делеція виявляється у 2/3 хворих. Синдром Прадера – Віллі розвивається, коли дитина успадковує делецію від батька, а причиною синдрому Ангельмана стає така ж делеція, одержана від матері. Таким чином, виникнення цих двох спадкових синдромів, що клінічно розрізняються, визначається тим, від кого з батьків було одержане хромосомне ушкодження.
Синдром Прадера – Віллі. Основними клінічними проявами хвороби є низький тонус м’язів, недорозвиток статевих органів, ожиріння, розумова відсталість, зменшені розміри кистей та стоп, а також множинні вади розвитку частин тіла. Частота захворювання складає 1:10000 – 1:20000 новонароджених незалежно від статі.
Нездужання починає проявлятися з перших днів життя дитини проблемами з годуванням через слабкість смоктального та ковтального рефлексів. Тримати голову хвора дитина починає після 6 місяців, сидіти – після року, а ходити на 3-4 році життя.
По досягненні 1,5-2 років м’язовий тонус міцніє, розвивається відчуття постійного голоду – хворий може їсти практично безперервно, внаслідок чого швидко розвивається тяжке ожиріння. Серед вад морфогенезу найбільш часто спостерігається видовжена голова, збільшена відстань між симетричними органами, епікант, мигдалеподібний розріз очей, «риб’ячий» рот і т.п. (Мал. 52).
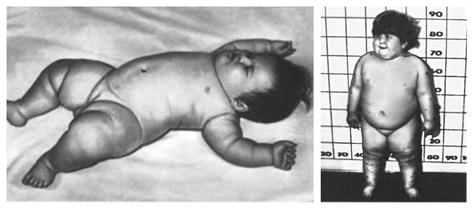
Мал. 52. Синдром Прадера – Віллі
Майже всі хворі на синдром Прадера – Віллі відзначаються розумовою відсталістю різного ступеня аж до крайньої імбецильності.
Лікування симптоматичне – дієта з обмеженням жирів та вуглеводів, гормональні препарати.
Тривалість життя хворих в середньому складає 25-30 років.
Синдром Ангельмана. Початкова назва цієї хвороби – синдром «щасливої ляльки» через характерні клінічні прояви: напади неконтрольованого сміху, різкі судомні рухи рук, незвичайна хода, плескання долонями і специфічний вираз обличчя. Частота її складає 1:20 000 новонароджених.
Основними клінічними проявами синдрому Ангельмана, крім визначених вище, є затримка розумового та фізичного розвитку (мала голова, пласка потилиця, велика нижня щелепа, надто широкий рот, рідкі зуби, часте висовування язика, недостатня пігментація шкірних покривів та волосся (мал. 53).

Мал. 53. Синдром Ангельмана
З віком у дитини більш помітними стають мовні вади, нервові розлади та розумова відсталість, яка може досягти рівня ідіотії.
Лікування симптоматичне.
Дата публикования: 2014-11-18; Прочитано: 935 | Нарушение авторского права страницы | Мы поможем в написании вашей работы!
