 |
Главная Случайная страница Контакты | Мы поможем в написании вашей работы! | |
Мітохондріальні хвороби. Мітохондріям належить провідна роль в утворенні енергії в результаті окислення вуглеводів, жирів та білків
|
|
Мітохондріям належить провідна роль в утворенні енергії в результаті окислення вуглеводів, жирів та білків. Дефект будь-якого з ферментів мітохондрій порушує їх функцію. При цьому в першу чергу страждають найбільш енергозалежні тканини і органи – центральна нервова система, скелетні та серцевий м’язи, нирки, печінка, ендокринні залози. На фоні хронічного дефіциту енергії в них рано чи пізно виникають патологічні зміни і розвиваються захворювання, які одержали назву мітохондріальних. Сучасній медицині відомо близько 200 таких хвороб. В їх клініці зустрічається сама різні патології, але домінують ураження центральної нервової системи та м’язової тканини.
Симптомами, типовими для мітохондріальних захворювань, є м'язові болі, слабкість і атрофія мускулатури, нездатність до фізичних навантажень, опущення верхніх повік, ушкодження периферійних нервових волокон, судоми, відсутність рефлексів, атрофія зорового нерва, нейросенсорна глухуватість, мігрень, летаргійні стани, порушення психомоторного розвитку (рухові акти, трудова діяльність, навички та вміння), недоумкуватість, розлад пам’яті, уваги, мислення, мови та поведінки.
Мутації, що виникли в мітохондріальних генах, передаються в нові мітохондрії при поділі цих органел. Виходить, що навіть у межах однієї клітини наявні мітохондрії з різними варіантами геномів. Таким чином, людина з мутацією в мітохондріальному гені несе суміш нормальної та мутантної ДНК. При цьому співвідношення мітохондрій з мутантними та нормальними геномами може бути будь-яким, тому прояв мітохондріальних захворювань у різних хворих неоднаковий. У подібних випадках мутації спочатку можуть взагалі не мати зовнішніх ознак. Нормальні мітохондрії до певного часу забезпечують клітини енергією, компенсуючи недостатність функції мітохондрій з дефектами. Це проявляється різним за тривалістю безсимптомным періодом при багатьох мітохондріальних захворюваннях. Проте рано чи пізно наступає момент, коли дефектні мітохондрії нагромаджуються в кількості, достатній для прояву патологічних ознак. Вік маніфестації захворювання варіює у різних хворих. Ранній початок захворювання приводить до тяжчого його перебігу та несприятливого прогнозу.
Успадкування мутацій у мітохондріальному геномі носить особливий характер. Якщо гени, локалізовані в ядерній ДНК, діти одержують порівну від обох батьків, то мітохондріальні гени передаються нащадкам тільки від матері. Це пов’язано з тим, що всю цитоплазму з мітохондріями, що містяться в ній, нащадки одержують разом з яйцеклітиною, тоді як в сперматозоонах цитоплазма практично відсутня. З цієї причини жінка з мітохондріальним захворюванням передає його всім своїм дітям, а хворий чоловік – ні.
Кожний із відомих сьогодні синдромів, викликаних порушенням функціонування мітохондрій, визначається якою-небудь мутацією наступного типу: нуклеотидні заміни в генах, втрати або вставки фрагментів мтДНК, зміна числа копій мтДНК.
Сьогодні ідентифіковано понад 20 спадкових мітохондріальних патологій, до яких належать спадкова атрофія зорових нервів Лебера, пігментний ретиніт (точніша назва хвороби: нейропатія, атаксія та пігментний ретиніт), міоклональна епілепсія з надзвичайно червоними м’язовими волокнами, нейросенсорна глухота тощо.
Атрофія зорових нервів Лебера. Відомо принаймні 10 точкових мутацій генів, які пов'язані з синдромом Лебера. Вони спричинюють заміну тієї чи іншої амінокислоти у одного з ферментів-дегідрогеназ, що призводить до порушення його активності. При даному захворюванні у людей 20-30 років відбувається майже повна втрата центрального зору через атрофію зорових нервів та дегенерацію гангліозного шару клітин сітківки. Хворіють переважно чоловіки (80-85%). Виявлено, що у 95% випадків причиною патології є мутації в трьох мітохондріальних генах – ND1 (мутація LHON 3460 A), ND4 (мутація LHON 11778 A) та ND6 (мутація LHON 14484 C) (мал. 46).
Решта 7 мутацій, пов’язаних з хворобою Лебера, вважаються «вторинними» і можуть підсилювати дію первинних мутацій, збільшуючи ризик прояву захворювання.
Синдром Лебера – найпоширеніше з усіх відомих на даний час мітохондріальних спадкових захворювань – його частота в Європі складає близько 1:25000.
Лікування здійснюється препаратами, які поліпшують обмін речовин, розширюють судини, та вітамінами, а також фізіотерапевтичними засобами (електростимуляція, магнітотерапія тощо).
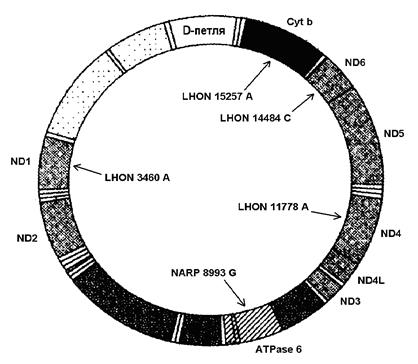
Мал. 46. Локалізація генів деяких мітохондріальних патологій
(М-хромосома)
Нейропатія, атаксія та пігментний ретиніт. Цю комплексну хворобу викликає точкова мутація NARP 8993 G в гені АТРase 6, який кодує один із ферментів АТФ-синтетазного комплексу. (мал. 46). Захворювання розвивається за наявності в клітинах 70-90% аномальної мтДНК. Ознаками патології є затримка розвитку, розумова відсталість, прогресуюче звуження зорових полів та нічна сліпота, біль та порушення чутливості у відповідних зонах іннервації, розлад координації довільних рухів і нейрогенна м'язова слабкість. До речі, сьогодні відомо принаймні 15 форм пігментного ретиніту (прогресуюче звуження зорових полів та нічна сліпота), спричинених домінантними та рецесивними мутаціями генів, які локалізовані в ряді аутосом та Х-хромосомі.
Якщо мутантна ДНК складає понад 90% від усієї мтДНК, то розвивається зовсім інша патологія – хвороба Лея, яка характеризується ураженням головного мозку, атрофією зорових нервів, зниженням тонусу м’язів, розладом координації довільних рухів, мимовільними ритмічними рухами очних яблук та обмеженням їх довільної рухомості.
Контрольні запитання до теми 5.4
1. Як розподіляються патології за типом успадкування?
2. Назвіть аутосомно-домінантні захворювання, які Ви знаєте.
3. Де локалізований ген, мутація якого призводить до хореї Гентинґтона, та в чому полягає ця мутація?
4. Назвіть характерні симптоми хореї Гентинґтона.
5. У якому віці проявляється хорея Гентинґтона та яка тривалість життя хворих?
6. Розкрийте генетичні причини синдрому Марфана.
7. Назвіть основні ознаки синдрому Марфана та тривалість життя хворих
8. Які найпоширеніші аутосомно-рецесивні патології Ви знаєте?
9. Поясніть загальні особливості успадкування аутосомно-рецесивних захворювань.
10. Розкрийте генетичні причини муковісцидозу.
11. Назвіть основні симптоми муковісцидозу.
12. Що Ви знаєте про генну терапію муковісцидозу?
13. Поясніть генетичний механізм фенілкетонурії.
14. Дайте коротку клінічну характеристику фенілкетонурії.
15. Розкрийте генетичні причини галактоземії.
16. Назвіть характерні ознаки галактоземії.
17. У чому полягає основна особливість успадкування Х-зчеплених патологій?
18. Охарактеризуйте Х-зчеплені домінантні патології на прикладі гіпофосфатемічного рахіту.
19. Охарактеризуйте Х-зчеплені рецесивні патології на прикладі м’язової дистрофії Дюшена.
20. Наведіть приклади Y-зчеплених захворювань. Яка характерна особливість їх успадкування?
21. Дайте коротку характеристику мітохондріальних патологій (у тому числі особливості успадкування) на прикладі атрофії зорового нерва Лебера.
Дата публикования: 2014-11-18; Прочитано: 1489 | Нарушение авторского права страницы | Мы поможем в написании вашей работы!
