 |
Главная Случайная страница Контакты | Мы поможем в написании вашей работы! | |
Строение двойного электрического слоя на границе электрод – раствор
|
|
При изучении электрокинетических и электрокапиллярных явлений были установлены определённые опытные закономерности. Корректная теория строения двойного электрического слоя металл – электролит должна давать их истолкование. Эти же факты служат критерием справедливости тех или иных вариантов теории двойного электрического слоя.
Наиболее важные результаты, полученные при изучении электрокинетических и электрокапиллярных явлений, сводятся к следующему.
1. Наряду с общим скачком потенциала на границе металл – электролит gM,L, входящим как зависящее от концентрации слагаемое в величину электродного потенциала Е, существует также электрокинетический, или z-потенциал, не совпадающий с общим скачком потенциала.
Обычно z-потенциал по абсолютной величине меньше gM,L- и Е-потенциала, и его зависимость от состава раствора более сложная. С увеличением концентрации электролита z-потенциал в большинстве случаев (если только в растворе нет поверхностно-активных ионов) уменьшается и стремится к нулю. При изменении концентрации раствора знак z-потенциала может меняться на обратный, хотя знак Е-потенциала остаётся тем же самым. Такое изменение знака z-потенциала и соответствующая ему перезарядка поверхности металла наблюдается в присутствии поверхностно-активных и поливалентных ионов.
2. Дифференциальная ёмкость, рассчитанная из электрокапиллярных кривых или измеренная непосредственно, зависит от потенциала электрода. Она имеет минимальное значение в области потенциалов, примыкающих к потенциалу незаряженной поверхности в данном растворе. Для растворов, не содержащих поверхностно-активных веществ, значение ёмкости в точке максимума электрокапиллярной кривой закономерно уменьшается по мере разбавления раствора. На отрицательной (нисходящей) ветви электрокапиллярной кривой ёмкость обычно меньше, чем на положительной (восходящей) ветви, и в растворах многих электролитов она мало изменяется с потенциалом.
Было предпринято несколько попыток создания отвечающих действительности представлений о строении двойного электрического слоя на границе между металлом и раствором; основные из них рассматриваются ниже.
Теория конденсированного двойного слоя Гельмгольца
Первую количественную теорию строения ДЭС на границе металл-раствор создал Гельмгольц (1853). По Гельмгольцу, ДЭС можно уподобить плоскому конденсатору, одна из обкладок которого совпадает с плоскостью, проходящей через поверхностные заряды в металле, другая - с плоскостью, соединяющей центры зарядов ионов в растворе, притянутых к поверхности металла электростатическими силами. Толщина двойного слоя l равна радиусу ионов r (см. рис. 22). По условию электронейтральности число притянутых к поверхности металла ионов должно быть таким, чтобы их заряды компенсировали поверхностные заряды металла, то есть
– qM = qL.
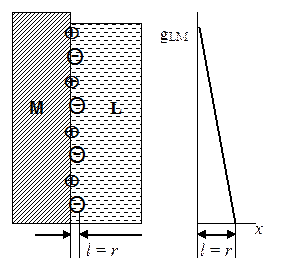
| Рис. 22. Модель ДЭС по Гельмгольцу: молекулярная картина и изменение потенциала с расстоянием от поверхности металла в глубь раствора |
Модель ДЭС, отвечающая этим простейшим представлениям, приводит к двум возможным значениям z-потенциала. Если все заряды, находящиеся в растворе, перемещаются вместе с жидкостью (при движении твердого тела относительно жидкости не увлекаются вместе с ним), то z-потенциал по величине будет совпадать с Е-потенциалом, и его изменение с концентрацией электролита должно подчиняться формуле Нернста. Если заряды, находящиеся в растворе, при относительном перемещении твердого тела и жидкости связаны только с твердым телом и перемещаются вместе с ним, то z-потенциал всегда равен нулю. Ни одно из этих следствий из теории Гельмгольца не согласуется с экспериментальными данными (если не считать, что z-потенциал может быть равен нулю в очень концентрированных растворах и при определенном составе раствора, отвечающем изоэлектрической точке). Теория Гельмгольца не объясняет также причины изменения заряда поверхности металла в присутствии ПАВ. Вместе с тем теория конденсированного двойного слоя позволяет получить значения емкости ДЭС, согласующиеся с опытом, и физически правдоподобную толщину ДЭС.
Таким образом, теория Гельмгольца в какой-то мере отражает истинную структуру ДЭС, но она не может истолковать многие опытные закономерности и должна рассматриваться лишь как первое приближение к действительности, нуждающееся в дальнейшем развитии.
Теория диффузного двойного слоя Гуи – Чапмана
В теории Гельмгольца не учитывается, что свойства ДЭС изменяются с концентрацией электролита и его температурой. Гуи (1910) и независимо от него Чапман (1913) попытались связать плотность заряда в ДЭС с составом раствора. Они учли, что помимо электростатических сил, возникающих между металлом и ионами, на ионы также действуют силы теплового молекулярного движения. При наложении этих двух сил ионы в растворе должны распределяться относительно поверхности металла диффузно - с убывающей при удалении от нее объемной плотностью заряда. Гуи и Чапман считали, что ионы можно рассматривать как материальные точки, не имеющие собственного объема, но обладающие зарядом, и что их распределение в поле заряда электрода подчиняется распределению Больцмана (см. рис. 23).
Величину удельного поверхностного заряда со стороны раствора находят так же, как плотность заряда ионной атмосферы по первому приближению теории Дебая и Гюккеля. В обоих случаях отправными уравнениями служат уравнения Больцмана и Пуассона. При определении достаточно использовать лишь одну координату – расстояние от поверхности электрода в глубь раствора. Это обстоятельство упрощало задачу и позволяло получить точное решение уравнения Пуассона – Больцмана. Теория Гуи – Чапмана была создана примерно за 10 лет до теории Дебая – Гюккеля и, вероятно, имела большое влияние на развитие последней.

| Рис. 23. Строение двойного слоя по Гуи – Чапману: молекулярная картина и изменение потенциала с расстоянием от поверхности металла в глубь раствора |
Теория Гуи – Чапмана лучше теории Гельмгольца согласуется с закономерностями электрокинетических явлений. Если предположить, что начиная с некоторого расстояния l 1 ионы уже не связаны прочно с поверхностью электрода при относительном перемещении твердой и жидкой фаз, то соответствующий этому расстоянию потенциал можно считать z-потенциалом (z< Е). При повышении концентрации раствора увеличивается плотность заряда qL, силы притяжения междуповерхностью металла и ионами другого знака заряда растут, а степень диффузности и z-потенциал уменьшаются, что согласуется с опытом. Однако теория не объясняет изменение знака z-потенциала и перезарядку поверхности с изменением состава раствора; знак z-потенциала по этой теории должен быть всегда таким же, как у общего скачка потенциала металл – раствор. Кроме того, теория Гуи – Чапмана оказывается менее удовлетворительной, чем теория Гельмгольца, при использовании ее для количественных расчетов емкости ДЭС: ни ход теоретической кривой дифференциальная емкость – потенциал, ни абсолютные значения емкостей не совпадают с опытными данными (количественный расчет емкости ДЭС приводит к величинам, которые на 7-10 порядков превышают опытные). Такое расхождение связано с тем, что теория Гуи – Чапмана не учитывает собственного объема ионов, которые отождествляются с материальными точками. В результате ничто не препятствует ионам в принятой модели подходить сколь угодно близко к поверхности металла.
Таким образом, теория Гуи – Чапмана оправдывается лучше всего там, где теория Гельмгольца оказывается неприложимой, и, наоборот, последняя дает лучшую сходимость с опытом в тех случаях, когда первая дает неверные результаты. Следовательно, строению ДЭС должно отвечать некоторое сочетание моделей, предложенных Гельмгольцем и Гуи – Чапманом. Такое предположение было сделано Штерном (1924) в его адсорбционной теории ДЭС.
Адсорбционная теория Штерна
Штерн полагал, что ДЭС состоит из двух частей: плотного и диффузного, разделенных плоскостью, называемой плоскостью Гельмгольца. Определенная часть ионов удерживается вблизи поверхности раздела металл – электролит, образуя гельмгольцевскую или конденсированную обкладку двойного слоя; толщина плотного слоя равна радиусу гидратированных ионов (0,3 – 0,4 нм), а его диэлектрическая постоянная значительно ниже, чем в объеме раствора. Это обусловлено довольно жесткой ориентацией диполей растворителя в плотном слое как под действием электрического поля электрода, так и в результате их взаимодействия с металлом. Остальные ионы, входящие в ДЭС, распределяются диффузно с постепенно убывающей плотностью заряда. Для диффузной части ДЭС Штерн, как и Гуи, пренебрег собственными размерами ионов; диэлектрическая постоянная диффузного слоя принималась равной диэлектрической постоянной растворителя в объеме раствора. Кроме того, Штерн высказал мысль, что в плотной части ДЭС ионы удерживаются за счет не только электростатических сил, но и сил специфической адсорбции, то есть силами некулоновского происхождения. Поэтому в растворах, содержащих поверхностно-активные ионы, их число в плотной части ДЭС может превосходить заряд поверхности металла на некоторую величину, зависящую от свойств ионов и заряда металла.
По Штерну, следует различать две модели ДЭС, одна из которых относится к растворам поверхностно-инактивных электролитов, другая - к растворам, содержащим специфически адсорбирующиеся ионы (см. рис 24).
В адсорбционной теории также сохраняется равенство:
- qM = qL = q1 + q2.
Плотность заряда со стороны раствора qL состоит из двух частей: плотности заряда в гельмгольцевском слое q1 и плотности заряда в диффузном слое q2 . Величина q2 зависит не от всего скачка потенциала между металлом и раствором, а лишь от той его части, которая приходится на падение потенциала в диффузной области ДЭС.
Теория Штерна позволяет определить z-потенциал как падение потенциала в диффузной части ДЭС, где уже потеряна прочная связь между металлом и ионами. При таком определении z-потенциал не должен совпадать с нерстовским потенциалом, как это и наблюдается на опыте. В растворах ПАВ z-потенциал не только не совпадает с общим скачком потенциала, но может отличаться от него и по знаку (см. рис. 24, б). Таким образом, теория Штерна смогла объяснить перезарядку поверхности твердого тела и изменение знака z-потенциала.
При бесконечно малой концентрации все заряды в растворе распределены диффузно, и строение ДЭС описывается теорией Гуи – Чапмана. Напротив, в концентрированных растворах строение ДЭС приближается к модели, предложенной Гельмгольцем. В области средних концентраций, где z сравним по величине с RT/F, его зависимость от концентрации можно выразить приближенными уравнениями:
z = В -  ln с (для положительных величин z),
ln с (для положительных величин z),
z = В¢ + ln с (для отрицательных значений z).
 а
а
|  б
б
|
| Рис. 24. Модель ДЭС по теории Штерна: а – для растворов поверхностно-инактивных электролитов, б – растворов, содержащих поверхностно-активные ионы |
Теория Штерна дает качественно правильную картину ДЭС. Определение емкости с использованием модели Штерна согласуется с опытом как по величинам емкости, так и по характеру ее зависимости от потенциала электрода и концентрации раствора. Но теория Штерна не свободна от недостатков. К их числу относится невозможность количественного описания емкостных кривых, особенно при удалении от потенциала нулевого заряда.
Дальнейшее развитие теории строения ДЭС
Современные теоретические представления о ДЭС базируются на основной модели Штерна, но содержат ряд усовершенствований, которые были внесены в теорию в последующие годы. В теории Штерна не учитывалось различие минимального расстояния, до которого могут приближаться к поверхности электрода электрические центры поверхностно-неактивных и специфически адсорбирующихся ионов. В действительности, ионы, которые специфически адсорбированы, частично дегидратированы со стороны металла, а потому они входят внутрь плотного слоя, и их электрические центры располагаются ближе к поверхности электрода, чем такие же центры полностью гидратированных поверхностно-неактивных ионов. В результате вместо одной плоскости Гельмгольца необходимо было ввести две плоскости: внутреннюю и внешнюю. На внутренней плоскости Гельмгольца локализуются центры специфически адсорбированных ионов, а внешняя плоскость Гельмгольца представляет собой границу максимального приближения к поверхности электрических центров всех ионов, находящихся в диффузной части ДЭС и участвующих в тепловом движении.
Было предпринято много попыток разработать теорию ДЭС, количественно согласующуюся с опытными данными (Райс, Фрумкин с сотр., Бокрис, Деванатхан, Есин, Мюллер, Парсонс, Эршлер и др.). Наибольшее признание получила модель Грэма (1947). Согласно Грэму, обкладка ДЭС, находящаяся в растворе, состоит не из двух, а из трех частей. Первая, считая от поверхности металла, называется внутренней плоскостью Гельмгольца; в ней находятся лишь поверхностно-активные ионы (заряд плоскости равен q1) либо, если их нет в растворе, молекулы растворителя (q1 = 0); потенциал ее, отнесенный к раствору, обозначается y1 . Следующая, удаленная от поверхности металла на расстояние, до которого могут подходить ионы (центры их заряда), называется внешней плоскостью Гельмгольца; ее общий заряд равен q2 , а потенциал плоскости y2 . За внешней плоскостью Гельмгольца располагается диффузный слой с потенциалом, изменяющимся от y2 до нуля и с плотностью заряда, совпадающей с q2 .
На рис. 25 дано схематическое изображение строения ДЭС по Грэму для незаряженной поверхности, заряженной положительно и отрицательно. В соответствии с допущением Грэма о том, что следует считаться лишь с поверхностной активностью анионов (в системах, не содержащих органических растворенных веществ), в первой плоскости Гельмгольца находятся только специфически адсорбирующиеся анионы, причем их поверхностная концентрация растет при переходе от незаряженной поверхности к заряженной положительно. При достаточно отрицательном заряде поверхности во внутреннем слое Гельмгольца остается лишь растворитель, и заряд его, так же как и в растворе, не содержащем ПАВ, делается равным нулю (q1 = 0). В этих условиях
qM = – qL = – q2
и ДЭС электрически эквивалентен двум последовательно включенным конденсаторам, то есть оправдывается формула, применимая и к модели Штерна:
 =
=  +
+ 
(С1 – емкость плотной части ДЭС, С2 – емкость диффузной части ДЭС).

| 
| 
|
| a | б | в |
| Рис. 25. Строение ДЭС металл – электролит по Грэму: а – поверхность металла не заряжена; б – поверхность металла заряжена положительно; в – поверхность металла заряжена отрицательно |
Грэм предполагал, что емкость конденсированной части ДЭС зависит от заряда металла, но не от концентрации раствора. Это предположение позволило Грэму предложить метод расчета кривых дифференциальной емкости для любых концентраций данного электролита, если имеется экспериментальная кривая хотя бы для одного его раствора. Проверка, проведенная Грэмом, показала хорошую сходимость рассчитанных кривых емкость – потенциал и кривых, полученных экспериментально.
Модель Грэма отражает основные черты и особенности структуры ДЭС металл – электролит. Однако и эта модель охватывает далеко не все аспекты проблемы:
1. Вряд ли допустимо рассматривать ионы, находящиеся во внутренней обкладке ДЭС, как равномерно размазанные по поверхности металла; надо учитывать дискретность зарядов.
2. В подавляющем большинстве случаев поверхность электрода поликристаллична и представляет собой набор участков, отвечающих граням разного индекса. Разные грани одного и того же металла обладают неодинаковыми работами выхода электрона и нулевыми точками; различия могут достигать нескольких десятых вольта. В итоге заряды граней оказываются не только неодинаковыми по значению, но могут оказаться и обратными по знаку. Таким образом, кристаллическая микронеоднородность может приводить к радикальным изменениям в структуре ДЭС.
3. Нерешенным остается вопрос, связанный с участием и вкладом электронов в структуру ДЭС.
Кроме разности потенциалов, создаваемой зарядами металла и ионами двойного слоя, электрические свойства границы раздела электрод/раствор зависят также от находящихся на поверхности диполей растворителя; ориентация диполей изменяется при изменении заряда электрода. Модели ДЭС, учитывающие изменение ориентации диполей растворителя, разрабатывались с начала 1960-х годов. В первых моделях предполагалось существование в плотной части ДЭС двух состояний диполей воды, ориентированных либо положительным, либо отрицательным концом к поверхности электрода. Эти диполи взаимодействовали с зарядами электрода и друг с другом по законам классической электростатики; при изменении заряда электрода изменялось долевое отношение диполей с противоположными ориентациями. В дальнейшем модели этого типа уточнялись с учетом значительного числа эффектов: возможного образования ассоциатов из двух или более молекул растворителя; наличия диполей, ориентированных параллельно или наклонно к поверхности; существования неэлектростатических взаимодействий между молекулами растворителя как в пределах ближайшего к поверхности монослоя, так и в соседних слоях.
Существенную роль в строении ДЭС на границе электрод/раствор играет выход электронного газа за пределы кристаллической решетки металла (ионного остова). Поверхностный потенциал металла c М(L) , измененный контактом с растворителем, также зависит от заряда электрода, что должно отразиться как на емкости ДЭС, так и на пограничном натяжении электрода. Вклад «металлической» составляющей в емкость ДЭС был впервые рассмотрен О.Райсом еще в 1928 г. Но эта работа значительно опередила свое время, и к обсуждению роли электронов металла в структуре и свойствах границы раздела электрод/раствор ученые всерьез приступили лишь после 1982 г.
Наконец, на строение границы электрод/раствор существенное влияние оказывает адсорбция нейтральных органических молекул. В этом случае раздвигаются обкладки двойнослойного конденсатора, снижается диэлектрическая постоянная плотного слоя, а также возникает дополнительный скачок потенциала, связанный с ориентированной адсорбцией органических диполей. Первая модель, описывающая обратимую адсорбцию на границе ртуть/вода простых алифатических соединений, была предложена А.Н.Фрумкиным еще в 1928 г. В основу этой модели, получившей название модели двух параллельных конденсаторов, были положены два уравнения, связывающие степень заполнения поверхности органическим веществом с его объемной концентрацией (изотерма адсорбции) и с зарядом электрода. Введение некоторых уточнений в эту модель позволило в дальнейшем с ее помощью описать весьма сложную форму кривых дифференциальной емкости на границе ряда металлов (Hg, Bi, Pb, Sn, Cd, Tl, Ga) с водными растворами многих органических веществ. В последние годы были предприняты усилия по разработке статистических теорий обратимой адсорбции органических молекул на поверхности электрода.
Дата публикования: 2014-11-19; Прочитано: 5891 | Нарушение авторского права страницы | Мы поможем в написании вашей работы!
