 |
Главная Случайная страница Контакты | Мы поможем в написании вашей работы! | |
Каталитический крекинг
|
|
Это процесс каталитического деструктивного превращения тяжелых фракций нефти в моторное топливо и сырье для нефтехимии, технического углерода и кокса. Процесс ведут на алюмосиликатном катализаторе при 480 – 530 оСи давлении 0,3 – 0,7 МПа.
9.3.1. Химические основы процесса. Основными реакциями каталитического крекинга являются:
1) расщепление высокомолекулярных углеводородов;
2) изомеризация;
3) дегидрирование циклоалканов в арены.
Реализация этих реакций приводит к получению карбюраторных топлив с высоким октановым числом, т. к. октановые числа изоалканов выше, чем у н-алканов, а у аренов – чем у циклоалканов и алканов.
9.3.2. Превращения алканов. Они подвергаются изомеризации и распаду на алканы и алкены меньшей молекулярной массы, чем исходные алканы. Первая стадия процесса – зарождение цепи – происходит по двум механизмам.
По первому – часть молекул алканов термически крекируется. Образовавшиеся алкены отрывают протоны от катализатора и превращаются в карбкатионы:
+
 RCH= CH2 + HA RCHCH3 + A-. (9.12 )
RCH= CH2 + HA RCHCH3 + A-. (9.12 )
По второму – образование карбкатиона происходит непосредственно из алкана путем отщепления от молекулы гидрид-иона под действием протонного центра или апротонного катализатора:
 RH + H+ R+ + H2 (9.13)
RH + H+ R+ + H2 (9.13)
или
 RH + L R+ + LH- (9.14)
RH + L R+ + LH- (9.14)
Так как отрыв гидрид-иона от третичного атома легче всего, изоалканы крекируются быстрее, чем н-алканы.
Термодинамика процесса такая, что, превращение например, карбкатиона С7Н15 + состоит в цепи ряда последовательно-параллельных реакций изомеризации и 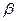 -распада. Химизм этого процесса представлен ниже.
-распада. Химизм этого процесса представлен ниже.
СН3(СН2)5СН2+


 изомеризация
изомеризация
 - распад +
- распад +
 СН3(СН2)4СН+СН3 СН3 – СН = СН2 + СН3(СН2)2СН2
СН3(СН2)4СН+СН3 СН3 – СН = СН2 + СН3(СН2)2СН2
 | |||
| |||
изомеризация - распад +
 СН3(СН2)3СН+СН2СН3 СН3 –СН2 - СН = СН2 + СН3СН2СН2
СН3(СН2)3СН+СН2СН3 СН3 –СН2 - СН = СН2 + СН3СН2СН2
 |
изомеризация - распад +
 СН3(СН2)3С+(СН3)2 СН2 = С(СН3)2 + СН3СН2СН2
СН3(СН2)3С+(СН3)2 СН2 = С(СН3)2 + СН3СН2СН2
Так как распад алкильных карбкатионов с образованием первичных и вторичных ионов С 1 – С 3 происходит труднее, чем с образованием третичных ионов с большим числом атомов углерода, то скорость крекинга растет с удлинением цепи. Например, в одинаковых условиях степень превращения составит, %: н- С 5 Н 12 – 1; н- С 7 Н 16 –3; н- С 12 Н 26 -18; н- С 16 Н 34 – 42.
Легкость распада ионов с отщеплением третичных карбкатионов водет к накоплению в продуктах распада алканов с семью и более атомами углерода.
Выделившиеся низкомолекулярные карбкатионы после изомеризации отрывают Н+ -ионы от исходного углеводорода и весь цикл реакций повторяется. Обрыв цепи происходит при столкновении карбкатиона с анионом катализатора: R+ + LH- RH + L.
В заключение следует отметить, что скорость реакций каталитического крекинга алканов на 1–2 порядка выше, чем при термическом крекинге.
9.3.3.Превращения циклоалканов. Скорость каталитического крекинга цикланов близка к скорости крекинга алканов с одинаковым числом атомов углерода. Для циклоалканов характерны следующие реакции:
1) дециклизация с образованием алкенов и диенов;
2) дегидрирование, приводящее к получению аренов;
3) изомеризация циклов и заместителей.
Глубина крекинга цикланов растет с увеличением числа заместителей в кольце (табл. 9.2).
Таблица 9.2
Зависимость глубины крекинга от заместителей в циклоалканах
| Молекула | Глубина крекинга, % | Молекула | Глубина крекинга, % |

|  СН3 СН3
  Н3С Н3С
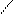 СН3 СН3
| 78,6 | |

  Н3С СН3 Н3С СН3
| 75,6 | 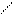 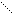  СН3
СН3 СН3
СН3
| 51,8 |
Возрастание глубины крекинга, как показано в табл. 9.2, связано с тем, что отщепление гидрид-иона с образованием одновременно карбкатиона протекает легче с третичным атомом углерода. В то же время гемструктуры (1,1-диметилциклогексан) отщепляют гидрид-ион от вторичного атома углерода, поэтому степень превращения в этом случае сопоставима со степенью превращения незамещенного циклогексана.
Распад циклогексил-иона идет двумя способами: 1) с разрывом связи
С – С; 2) с разрывом связи С – Н.
При разрыве связи С – С образуются алкены и алкадиены следующим образом:
R



 R +
R +


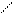

 + CH2 = C – (CH2)3CH2. (9.15)
+ CH2 = C – (CH2)3CH2. (9.15)
Алкенильный ион далее изомеризуется в аллильный:
R R
+ +
 CH2 = C – (CH2)3CH2 CH2 = C – СН(CH2)2CH3. (9.16)
CH2 = C – (CH2)3CH2 CH2 = C – СН(CH2)2CH3. (9.16)
Далее процесс развивается так:
 |

 R R
R R
+ +
 CH2 =C–СН(CH2)2CH3 +R/H CH2 = C –СН(CH2)3CH3 + R/ (9.17)
CH2 =C–СН(CH2)2CH3 +R/H CH2 = C –СН(CH2)3CH3 + R/ (9.17)
или происходит передача протона алкену или катализатору:
+
 CH2 =C(R)–СН-(CH2)2CH3 + R//CH = CH2
CH2 =C(R)–СН-(CH2)2CH3 + R//CH = CH2

 +
+
 CH2 =C(R)СН =CHCH2CH3 + R//CHCH3. (9.18)
CH2 =C(R)СН =CHCH2CH3 + R//CHCH3. (9.18)
|
Распад циклогексил-карбкатиона с расщеплением связи С – Н энергетически более выгоден, так как через промежуточные циклоалкены и циклоалкины образуются арены:
+ R R

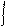

 С – H
С – H  (9.19)
(9.19)
- H
Циклоалкены подвергаются крекингу быстрее, чем циклоалканы:
R R R R + R/




 -H+ - Н+ - H+ R/ (9.20)
-H+ - Н+ - H+ R/ (9.20)
 CH + C + - RH+.
CH + C + - RH+.
H Н
Выход аренов составляет 25 % и более от суммы продуктов превращения циклогексанов, а газовая фаза содержит повышенное количество водорода по сравнению с газами крекинга алканов.
Имеет место также изомеризация циклогексанов в циклопентаны и обратно:



 +
+


 НС
НС


 С – Н + + (9.21) Н+ СН3 СН3
С – Н + + (9.21) Н+ СН3 СН3
Эта реакция протекает через протонированное пропановое кольцо.
Равновесие в этой реакции сильно сдвинуто вправо, так как в условиях каткрекинга циклопентаны более устойчивы, чем циклогексаны. Но циклогексаны дегидрируются в арены и тем самым равновесие сдвигается влево. Вероятность превращения циклогексана в тот или иной продукт в итоге зависит от катализатора.
Если в молекуле циклоалкана длинные боковые цепи, то возможна изомеризация боковой цепи и /или деалкилирование.
Бицикланы ароматизируются в большей степени, чем моноциклы. Декалин превращается в нафталин на 33 % при 500 оС, тетралин – на 88 %.
9.3.4. Превращения алкено в. Скорость их каткрекинга на 2 – 3 порядка выше соответствующих алканов и циклоалканов, что объясняется термодинамикой образования из них карбкатионов:
+
Н2С = СНСН2СН3 + Н+ Н3ССНСН2СН3 + 757 кДж/моль. (9.22)
Вместе со многими общими реакциями с алканами алкены обладают и специфическими реакциями перераспределения водорода и циклизации.
Сущность таких реакций состоит в том, что в присутствии кислых катализаторов часть алкенов теряет водород и образуются полиненасыщенные углеводороды, а другая часть алкенов гидрируется выделившимся водородом и переходит в алканы, например:
2С8Н16 С8Н14 + С8Н18. (9.23)
При этом сильноненасыщенные углеводороды полимеризуются, дегидрируются и, теряя водород, превращаются в кокс.
Циклизация алкенов может привести к образованию циклопентанов, циклопентенов и аренов по схеме:
СН3(СН2)3СН = СН2
|
 |
- Н2 - 2Н2 -3Н2


 СН3 СН3 СН3
СН3 СН3 СН3
 | |
метилциклопентан метициклопентен метилциклопентин бензол
Пятичленные циклы изомеризуются в шестичленные и далее ароматизуются.
9.3.5.Превращения аренов. Незамещенные арены при каталитическом крекинге стабильны. Монометилзамещенные арены по реакционной способности сравнимы с алканами, арены с более длинными алкильными заместителями по стабильности близки к алкенам. Основной реакцией для них является деалкилирование. Это объясняется тем, что ароматическое кольцо обладает большим сродством к протону по сравнению с алкил-ионом:
H



 +
+



 (СН2)2R + H+ (CH2)2R +CH2CH2R (9.24)
(СН2)2R + H+ (CH2)2R +CH2CH2R (9.24)
в свою очередь:
 +
+

 CH2CH2R CH2 = CHR + Н+. (9.25)
CH2CH2R CH2 = CHR + Н+. (9.25)
Скорость реакции возрастает с увеличением длины алкильного заместителя, а также в ряду:
С6Н5 – Rперв. < С6Н5 – Rвтор. < С6Н5 – Rтрет.
Именно такой ряд обусловлен различной устойчивостью образующихся карбкатионов.
Для метилзамещенных аренов отщепление карбкатиона затруднено, и протекают в основном реакции диспропорционирования:
 2С6Н5СН3 С6Н6 + С6Н4(СН3)2 (9.26)
2С6Н5СН3 С6Н6 + С6Н4(СН3)2 (9.26)

 и изомеризация по положению заместителя: СН3 Н3С СН3 Н3С (9.27).
и изомеризация по положению заместителя: СН3 Н3С СН3 Н3С (9.27).
СН3 СН3
Полициклические арены сорбируются на катализаторе и подвергаются постепенной деструкции и перераспределению водорода с образованием кокса.
Представляет интерес сравнение механизмов реакций углеводородов при термическом и каталитическом крекингах (табл. 9.3).
Таблица 9.3
Сравнение механизмов термического и каталитического крекинга
| Углево-дороды | Термический крекинг | Каталитический крекинг |
| Алканы | Расщепление по С-С связи (в газах преобладают углеводороды С1-С2).Дегидрирование низших алканов. | Расщепление по С-С – связи (в газах преобладают углеводороды С3-С4). Дегидрирование. |
| Алкены | Расщепление по С-С - связи. Дегидрирование. Полимеризация. Диеновый синтез. | Расщепление по С-С связи. Дегидрирование. Перераспределение водорода. Циклизация. |
| Диены | Диеновый синтез | Полимеризация с перераспределением водорода и образованием смолы и кокса, а также алканов. |
| Цикло-алканы | Расщепление по С-С - связи. Дегидрирование. Ароматизация | Расщепление по С-С с вязи. Дегидрирование. Ароматизация. Изомеризация |
| Арены | Конденсация с образованием многоядерных аренов. Расщепление алкилбензолов по -связи С – С.
| Расщепление алкилбензолов по  -связи С – С. Изомеризация. -связи С – С. Изомеризация.
|
9.4. Катализаторы каталитического крекинга.
Современные катализаторы каталитического крекинга представляют собой цеолитсодержащие алюмосиликаты, состоящие на 10-25 % из цеолита типа Y в редкоземельной или декатионированной форме, равномерно распределенного в аморфном алюмосиликате в виде микросферической или шариковой форм.
Природные цеолиты представляют собой кристаллические алюмосиликаты с общей формулой Me2/nO·Al2O3·xSiO·kH2O, где Ме – преимущественно Na, Ca, K, Ba и др.; х – силикатный модуль. Для цеолита типа А он равен 1,8 – 2,3, типа Х 2,3 – 3,0, типа Y 3,0 - 6,0.
Промышленные цеолиты отличаются от природных тем, что при их подготовке из них удаляют воду и заменяют путем ионного обмена катионы металлов на катионы водорода или аммония NH4 +. Такие катализаторы обладают активностью на 2 – 3 порядка выше, чем природные цеолиты.
Структура цеолита образована тетраэдрами SiO 2 и AlO 4- . Одиночный отрицательный заряд алюминат-иона компенсируется находящимися в пустотах кристаллической решетки цеолита катионами металла. Цеолиты с одновалентными катионами металлов (Na +, K +, Li + и др.) неактивны. Замена Ме + на Ме 2+ или Ме 3+ приводит к декомпенсации зарядов и создает высокую напряженность электростатического поля, достаточную для образования карбкатионов в результате смещения электронной пары:
 С – Н С+ - Н- или С = С С- - С+.
С – Н С+ - Н- или С = С С- - С+.
Аморфный алюмосиликат, в котором распределен цеолит, обладает собственной активностью. Каталитически активными центрами алюмосиликатов являются кислоты Бренстеда или Льюиса.
В качестве кислоты Бренстеда может быть протон, образующийся из воды и хемосорбированный ненасыщенным атомом алюминия (а), протон гидроксильной группы, связанный с атомом алюминия (б) или кремния (в):
| | | |


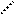 O O O O H+ - О – Si – Al: O - Si – O –Al-:. H+
O O O O H+ - О – Si – Al: O - Si – O –Al-:. H+
H
O O O O
а) б)
О –




 -
-
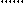 - О – Аl – O – Si – O H +
- О – Аl – O – Si – O H +
 O O
O O
в)
Апротонным кислотным центром может быть также структура:
O O
| |
– O – Si – Al – O –.
|
O
Переход протонной кислотности в апротонную описывается схемой:

 O O
O O
| |
- O – Al- – O – H H+ H2O + - O – Al
O O.
Наибольшее значение имеют протонодонорные центры, т. к. дегидратированный алюмосиликат неактивен. В цеолитсодержащих алюмосиликатных катализаторах роль катиона металла состоит в увеличении подвижности протона и стабильности кислотных центров Бренстеда, а также созданием дополнительного количества кислотных центров протонизацией воды:

 Ме п + + Н2О Ме п + ОН- Н+
Ме п + + Н2О Ме п + ОН- Н+
Именно поэтому скорость реакции на таком катализаторе на 2–3 порядка выше, чем на аморфном. Цеолиты обладают также очень высокой термической и механической стабильностью.
Дата публикования: 2014-11-02; Прочитано: 2503 | Нарушение авторского права страницы | Мы поможем в написании вашей работы!
