 |
Главная Случайная страница Контакты | Мы поможем в написании вашей работы! | |
Тема 3. Теории элементарного акта химической реакции
|
|
Лекция 4-2015
Обычно нестационарный режим является нежелательным, особенно в крупномасштабном производстве, поэтому наиболее распространенными являются проточные реакторы, в которых процессы тепло- и массообмена легко регулируются. В кинетическом анализе обычно используются два типа модельных реакторов: идеального перемешивания и идеального вытеснения. В проточном реакторе, в отличие от периодического, появляется свой масштаб времени, не зависящий от астрономического времени. Время пребывания в проточном реакторе τ равно отношению объема реакционного пространства к объемной скорости потока
τ = VR/QA.
Для реактора идеального вытеснения, справедливы кинетические уравнения, указанные в табл. 2, при использовании вместо времени t времени пребывания t = VR/QA. Покажем это на примере реакции 1-го порядка А ® В, протекающей в цилиндрическом реакторе идеального вытеснения.
dVR


 QA
QA
 C(0)
C(0)
Рис. 13. Цилиндрический реактор идеального вытеснения: QA – объемная скорость потока, м3/с; dVR – элемент объема реактора, в котором превращается часть вещества dx.
 В стационарном режиме в элементе объема накопление вещества не происходит, т.е.
В стационарном режиме в элементе объема накопление вещества не происходит, т.е.
 (2.25)
(2.25)
откуда rdVR= QAC(0)dx или, разделяя переменные, получим
dVR/QA = C(0)dx/r (2.26)
Левая часть уравнения (26) это дифференциал от времени пребывания t, поэтому с учетом начального условия: при t =0, x = 0 найдем общее выражение связи времени пребывания со степенью превращения для реакции, протекающей в реакторе идеального вытеснения:
 (2.27)
(2.27)
Ясно, что для решения этого уравнения необходимо выразить скорость реакции как функцию степени превращения реагента. Проиллюстрируем решение (27) на примере реакции 1-го порядка
r = kCA = kC(0)(1-x) (2.28)
Затем подставим (28) в (27), и после взятия интеграла, получим выражение
k.t = -ln(1-х) (2.29)
аналогичное уравнению, приведенному в табл. 2.
Рассмотрим реакцию 1-го порядка, протекающую с изменением числа молей:
А ® В + D
и выразим концентрацию реагента через конверсию:
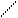
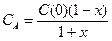 (2.30)
(2.30)
((1+x) в знаменателе появляется из-за увеличения числа молей в реакционной смеси по мере протекания реакции). Подставляя уравнение (30) в выражение скорости реакции, возьмем интеграл (2.27), который в этом случае имеет вид:
k.t = -2ln(1-х) – х (2.31)
Рассмотрим теперь кинетику реакции, протекающей в реакторе идеального перемешивания. Реактор идеального перемешивания является безградиентным, т.е. скорость процесса в реакторе не зависит от тепло- и массопереноса, геометрии реактора, скорости потока и др. макроскопических параметров.
  С С
 C(0) C(0) 
 С1 t1
С2 t2
Объем реактора С1 t1
С2 t2
Объем реактора
| Рис. 14. Профиль концентрации реагента в объеме реактора идеального перемешивания: C(0) начальная, С1 и С2 текущая концентрация реагента; t2>t1 |

В реакторе идеального перемешивания концентрация исходного вещества уменьшается скачком на входе реакционной смеси в реактор, а смесь на выходе из реактора имеет тот же состав, что и в любой точке внутри реактора. В таком реакторе расчеты существенно упрощаются по причине отсутствия градиентов концентраций – следовательно, не надо интегрировать дифференциальные уравнения. Запишем уравнение материального баланса по исходному веществу А
QA(C0 – CA) – rVR = 0 (2.32)
или вводя время пребывания в реакторе
C0 – CA = τ.rA (2.33)
Разделив (33) на C0, получим
x = τ rA/C0 или τ = x C0 / rA (2.34)
Для реакции 1-го порядка rA= kCA кинетическое уравнение в реакторе идеального перемешивания примет конкретный вид:
 или
или  (2.35)
(2.35)
или можно записать выражение для конверсии реагента:
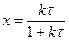 (2.36)
(2.36)
Видно, что зависимость х(τ) не содержит величины концентрации, как и в случае протекания реакции в реакторе вытеснения и в статической системе. Таким образом, для реакции 1-го порядка кривые х(τ) (или х(t)) инвариантны не только к концентрации исходного вещества, но и к типу реактора.
Тема 3. Теории элементарного акта химической реакции
Все рассмотренные выше кинетические уравнения направлены на определение констант скорости по экспериментальным данным С(τ) или х(τ). В свою очередь, константа скорости представляет собой основную величину, необходимую для проведения любых кинетических и технологических расчетов. Экспериментальное определения констант является достаточно трудоемкой задачей, даже в случае относительно простых реакций. Поэтому была поставлена задача расчета константы скорости реакции, исходя из теоретических представлений, как в термодинамике для констант равновесия. В теоретической кинетике были предложены 2 подхода:
1) теория активных соударений и
2) теория переходного состояния (абс. скор. реакции).
3.1 Теория активных соударений (АС) была разработана Аррениусом и Хиншельвудом в развитие 1-го и 2-го постулатов кинетики (ПК). В теории АС реагирующие частицы упрощенно считаются упругими шарами. Поскольку для протекания реакции необходимо столкновение 2 частиц (1й ПК), суммарная энергия которых ≥ Еа (2й ПК), для расчета скорости реакции необходимо рассчитать число столкновений (z) в единицу времени, в единице реакционного пространства и найти долю активных соударений (ε). Произведение zε будет представлять собой расчетную скорость реакции. Активными считаются соударения, в которых система частиц имеет энергию Е≥Еа.
В качестве реакционного пространства выберем так называемый цилиндр соударений с диаметром (δAB), равным полусумме диаметров молекул А и В, и длиной, равной длине свободного пробега (l0) между двумя столкновениями. Очевидно, что l0 равна произведению средней скорости движения молекул (ω) на время между двумя столкновениями Δt.
l0=ωΔt






 А
А
А В В δAB = ½(δA +δB)
Рис. 16. Цилиндр соударений молекул A и B
Выразим скорость движения молекул согласно кинетической теории газов
ω = [8RTμ/π] 0,5 (3.1)
где μ = (Ма + Мв)/Ма•Мв – функция молекулярных масс реагентов. Тогда число столкновений молекул А и В в единицу времени Δt, в единичном цилиндре определяется выражением:
z0 = 5,01•δAB2 [RTμ] 0,5 (3.2)
Величина z0 при единичных концентрациях А и В имеет порядок 1011 (М•с)-1. Полное число соударений в единицу времени в единице реакционного пространства равно Z = z0•nAnB, где nA и nB число молекул реагентов в указанном пространстве. Для расчета числа соударений молекул А с молекулами В и перехода к концентрации надо умножить Z на число Авогадро NA.
Доля активных соударений определяется функцией распределения молекул по энергии Максвелла-Больцмана, согласно которой
ε =  = exp(-Ea/RT) (3.3)
= exp(-Ea/RT) (3.3)
Заметим, что доля активных соударений растет с температурой существенно быстрее, чем кинетическая энергия частиц. (Например, при увеличении температуры от 500 до 800К средняя кинетическая энергия частиц увеличивается в 1,6 раза (как Т), а доля молекул с энергией
Е≥ 60 кДж/моль возрастает больше, чем в 1,7 млн).
Таким образом, перемножая уравнения 2 и 3, получим расчетное выражение скорости реакции по теории АС:
r = 5,01CACBNA δAB2 [RTμ] 0,5·exp(-Ea/RT) (3.4)
При сопоставлении (3.4) с уравнением кинетики видно, что величина константы скорости по теории АС имеет вид:
k = 5,01NA δAB2 [RTμ]0,5·exp(-Ea/RT), (3.5)
где А = 5,01NA δAB2 [RTμ] 0,5
Теория АС позволяет рассчитать только величину k, а Еа определяют экспериментально.
Посмотрим, насколько хорошо «работает» теория активных соударений. С этой целью сопоставим экспериментальные и расчетные значения предэкспоненциального множителя (f) для некоторых газофазных реакций в табл. 3.
Таблица 3. Проверка применимости теории АС для газофазных бимолекулярных реакций
| Реакция | А, (М·с)-1 | Е, кДж/моль | f =Aэ/Aт | |
| Эксперимент | Теория | |||
| 1) 2NOCl ® 2NO + Cl2 | 9,4·109 | 5,9·1010 | 0,16 | |
| 2) Н2+СН3· ®Н· + СН4 | 6·109 | 7,5·1011 | 1,20 | 0,008 |
| 3) С2Н4 + Н2 ® С2Н6 | 1,2·106 | 7,3·1011 | 1,7·10-6 | |
| 4) K + Br2 ® KBr + Br | 1,0·1012 | 2,2·1011 | 0,0 | 4,8 (К+) |
| 5) 2HJ ® H2+ J2 | 4,1·1010 | 9,2·1010 | 178,2 | 0,45 |
Как видно из табл. 3, теория АС для реакций 1, 4, 5 дает качественное соответствие предэкспоненциального множителя в пределах порядка величины, а для реакций 2 и 3 результативными являются лишь 0,008 и 1,7 из миллиона соударений. Следовательно, имеется неучтенный фактор протекания реакции. Этот фактор, названный стерическим, учитывает вероятность благоприятной конфигурации частиц при соударении. Например, для протекания реакции 2 необходимо, чтобы ось молекулы водорода Н-Н при соударении была перпендикулярна плоскости метильного радикала, а для реакции 3 необходимо, чтобы оси молекул С2Н4 и Н2 были параллельны (для максимального перекрывания s–орбитали водорода c π–орбиталью этилена).
Таким образом, согласно уточненной теории АС скорость реакции определяется тремя факторами: стерическим (f), энергетическим (ε) и транспортным (Z) или
r = f.ε. Z (3.8)
причем 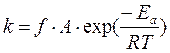 (3.9)
(3.9)
Здесь транспортный фактор Z определяет частоту соударений молекул реагентов, энергетический фактор ε учитывает долю молекул с энергией, необходимой для реакций, а стерический фактор учитывает долю молекул с надлежащей конфигурацией при соударении.
Очевидная недостаточность теории АС стимулировала поиск более перспективных моделей расчета скорости химических реакций. Такая модель активированного комплекса была положена в основу теории абсолютных скоростей химических реакций.
3.2 Теория переходного состояния (ПС) была разработана Эйрингом и Поляни (1935) с использованием аппарата статистической термодинамики. В модели переходного состояния предполагается, что исходная конфигурация реагирующих частиц, обладающих достаточной энергией для преодоления барьера реакции, преобразуется при непрерывном изменении межатомных расстояний и соответствующей энергии системы в активированный комплекс, который обязательно превращается в продукты реакции. Например, для реакции
АА + В ® {AAB} à ABA
для превращения исходных молекул АА и В в продукты они обязательно должны образовать активированный комплекс ААВ (рис. 16). В этом комплексе {AAB} старые связи существенно искажены и наметились уже новые связи, однако их конфигурация и энергия не соответствуют стабильным молекулам.
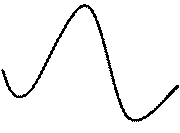
В соответствии с теорией ПС динамику превращения молекул В и АА в продукт АВА можно проиллюстрировать на рис. 17, где приведены результаты расчета изменений длины связей и соответствующей энергии системы.
Как видно из рисунка, при сближении частиц (пунктир) они преодолевают силы отталкивания (например, за счет кинетической энергии). При этом потенциальная энергия системы увеличивается до максимальной (точка M*▲), то есть, наивысшей точкой перехода между «потенциальными ямами», где находятся стабильные молекулы АА и АВА. Следует отметить, что пунктирная трасса М1 à М* àМ2, соответствует минимальным затратам энергии: для других маршрутов реакции требуются еще большие затраты энергии.
 Рис. 17. Карта поверхности потенциальной энергии системы в ходе реакции
АА + В ® ABA, кривые – изоэнергетические линии; точки ■,▲и ●
соответствуют исходным реагентам, комплексу {AAB} и продукту
Рис. 17. Карта поверхности потенциальной энергии системы в ходе реакции
АА + В ® ABA, кривые – изоэнергетические линии; точки ■,▲и ●
соответствуют исходным реагентам, комплексу {AAB} и продукту
|
Энергетическая развертка движения системы по пути минимального действия, приведенная на рис. 18, является двумерным представлением трехмерной диаграммы (рис. 17) зависимости энергии системы реагирующих частиц от их конфигурации, причем точка М является седловиной на перевале между минимумами потенциальной энергии для стабильных молекул. Этой точке соответствует активированный комплекс {AAB}, который можно рассматривать как промежуточное соединение, обладающее некоторой энергией и набором состояний по всем степеням свободы.
Е {AAB}*
  ▲
δ
АА+
В
АВА
■ ▲
δ
АА+
В
АВА
■
 ●
Координата реакции, ρ ●
Координата реакции, ρ
| Рис. 18. Изменение потенциальной энергии в ходе превращения исходных веществ АА и В в продукт ABA через активированный комплекс {AAB} |
Здесь термин «путь или координата реакции» относится не к пространственному перемещению комплекса, а к движению его фигуративной точки по энергетической поверхности. Участок δ на вершине потенциального барьера активированный комплекс проходит за время τ – или среднее время жизни АК.
Рассматриваемый линейный трехатомный активированный комплекс (n= 3) имеет 3х3 = 9 степеней свободы: 3 поступательных (по осям x-y-z), 2 вращательные (вокруг оси А-А-В и вокруг центрального атома А) и 4 колебательных (2 валентные и 2 деформационные) степеней свободы.
В теории ПС предполагается, что активированный комплекс находится в состоянии равновесия с исходными реагентами, причем одна из колебательных степеней свободы вырождается в поступательную, по координате ρ, и далее {AAB}* необратимо превращается в продукты:
K k
А-А + В «{AAB}* à A-B-A
В соответствии с теорией, скорость реакции будет равна числу активированных комплексов, проходящих через потенциальный барьер вдоль координаты реакции в единицу времени, в единице реакционного пространства. Следует также учесть вероятность прохода через барьер со скоростью u*, введя так называемый трансмиссионный коэффициент χ:
r = χCК*/τ или r = χCК*u*/δ (3.10)
С другой стороны, скорость бимолекулярной реакции можно выразить, используя 1й постулат кинетики:
r = kCАCВ (3.11)
или, исключая скорость из уравнений 10 и 11, можно найти величину константы:
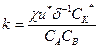 (3.12)
(3.12)
В этом уравнении относящиеся к АК величины u*, δ и CК* нельзя определить экспериментально, но можно выразить через определяемые величины, используя аппарат статистической термодинамики для равновесных процессов. Так, концентрацию активированного комплекса найдем из условия равновесия:
CК* = К*САCВ (3.13)
и получим выражение
k = χu*δ-1К* (3.14)
Скорость прохода активированного комплекс через барьер, выразим согласно молекулярно-кинетической теории:
 (3.15)
(3.15)
Выделим поступательную сумму по состояниям из выражения константы равновесия
QП*= δh-1(2πm*RT/NA)1/2, (3.16)
где h – постоянная Планка, и определим константу равновесия КС* без одной поступательной степени свободы как
Кс* = К*/QП* (3.17)
Подставим (3.15) - (3.17) в (3.14) и получим искомое выражение для константы скорости:
k =χRTh-1NA-1Кс* (3.18)
Для уточнения физического смысла используемых ранее терминов энергии активации, предэкспоненциального множителя k0, и стерического фактора преобразуем (3.18), выразив константу равновесия через энергию Гиббса
 , а энергию Гиббса - в виде
, а энергию Гиббса - в виде
∆Go* = ∆Ho* - T∆So*, тогда:
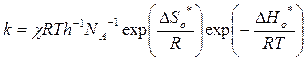 (3.19)
(3.19)
Это основное уравнение теории активированного комплекса.
Из уравнения (19) видно, что величина предэкспоненциального множителя
k0=χRTh-1NA-1.exp(∆So*/R) (3.20)
зависит от частоты колебаний активированного комплекса и изменения энтропии при его образовании.
Для нахождения связи между энергией активации и теплотой активации АК (∆Ho*) прологарифмируем (3.19)
 и возьмем производную по температуре:
и возьмем производную по температуре:
 (3.21)
(3.21)
Как видно из (3.21) энергия активации реакции Е равна сумме энтальпии активации активированного комплекса и RТ:
Е = ∆Ho*+ RТ (3.22)
Уравнение теории АК можно использовать для расчета предэкспоненциального множителя k0 в случае простых реакций (или оценки для сложных реакций). Из (3.19) видно, что стерический фактор отражает изменение энтропии при образовании АК
f~ exp(∆So*/R) (3.23)
.
По теории АК можно оценить величину стерического фактора и для реакций сложных молекул:
f= (Qкол /Qвращ)5 (3.24)
Обычно для колебательной энергии сумма по состояниям ~ 1, а для вращательной энергии сумма по состояниям составляет от 10 до 100. Это объясняет низкие значения стерического фактора 10-5 – 10-10, наблюдаемые экспериментально.
Можно получить информацию о механизме реакции. С этой целью в эксперименте определяют значения Еа, k и, используя уравнение (3.19), рассчитывают ∆So*. Если по расчету ∆So*< 0, это означает, что активированный комплекс является более упорядоченным, чем исходные реагенты. В случае ∆So*> 0, наоборот, следует принимать во внимание диссоциативные процессы при формировании АК.
Дата публикования: 2015-11-01; Прочитано: 742 | Нарушение авторского права страницы | Мы поможем в написании вашей работы!
