 |
Главная Случайная страница Контакты | Мы поможем в написании вашей работы! | |
Молярна концентрація еквівалента або нормальність (СН) визначається
|
|
відношенням кількості розчиненої речовини еквівалента ( ) до об’єму розчину (V), моль/л:
) до об’єму розчину (V), моль/л:
 |
 |
Моляльна концентрація (Cm) визначається відношенням кількості розчиненої речовини (
 ) до маси розчинника (
) до маси розчинника (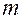 ), моль/кг:
), моль/кг:
 |
Масова концентрація (або титр) Т визначається відношенням маси роз-чиненої речовини (
) до об’єму розчину (V), г/мл:
Розрізняють такі частки розчиненої речовини: молярну, масову та об’єм-ну. Найчастіше вживаються масова та молярна частки.
Масова частка w або процентна концентрація С% визначається відно-шенням маси розчиненої речовини до маси всього розчину:

Молярна частка розчиненої речовини або мольна частка (N) визначається відношенням числа молів розчиненої речовини до сумарного числа молів усіх компонентів розчину:
 або в загальному випадку
або в загальному випадку 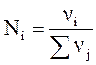 .
.

3.2. ЗАГАЛЬНІ ВЛАСТИВОСТІ РОЗЧИНІВ
Властивості розчину завжди відрізняються від властивостей кожного з його компонентів. Ця зміна властивостей зумовлена, з одного боку, характером взаємодії між компонентами, а з іншого — зменшенням концентрації молекул кожної з речовин під час розподілу в ній молекул іншої речовини.
Розведені розчини, в яких ефекти взаємодії розчиненої речовини з роз-чинником незначні, мають властивості, які залежать тільки від кількості роз-чиненої речовини в розчині і не залежать від їхньої природи. Такі властивості називаються колігативними. До них належать: тиск пари розчинника над роз-чином, температура замерзання (кристалізації) розчину та температура кипіння розчину, осмотичний тиск розчину.
Тиск пари над розчином. Кожна рідина характеризується сталою величиною тиску насиченої пари над поверхнею за даної температури. Наявність на поверхні (як і в об’ємі) розчину часточок нелеткої розчиненої речовини ускладнює випаровування розчинника. Тому тиск насиченої пари розчинника над розчином (Р) завжди менший, ніж над чистим розчинником (Р0) (Рис. 6).
Різницю між цими величинами називають зниженням тиску пари над розчином. Відношення величини зниження тиску пари над розчином до тиску насиченої пари над розчинником називають відносним зниженням тиску пари над розчином
 .
.
Залежність зниження тиску пари над розчином від концентрації розчиненої речовини описується першим законом Рауля: відносне зниження тиску насиченої пари розчинника над розчином дорівнює мольній частці розчиненої речовини:


де N2 — мольна частка розчиненої речовини;
n2 — число молів розчиненної речовини;
n1 — число молів розчинника.
 |
Р, кПа
 |
Рис.6. Залежність тиску насиченої водяної пари від температури для твердої (1), рідкої (2) води та для розчину (3)
Температура кипіння та замерзання розчинів прямо пропорційно залежить від тиску насиченої пари розчинника над розчином нелеткої речовини.
Температурою кипіння рідини є температура, за якої тиск її насиченої пари стає таким, що дорівнює зовнішньому тиску (рис.6). Оскільки за заданої температури тиск над розчином завжди менший ніж над розчинником, розчин закипає при більш високій температурі ніж розчинник.
Температурою замерзання рідини є температура, за якої тиск насиченої пари цієї рідини над її рідкою фазою стає таким, що дорівнює тиску пари над її твердою фазою. Оскільки над розчином тиск пари розчинника менший, ніж над чистим розчинником, розчин замерзає при більш низькій температурі, ніж роз-чинник.
 |
За другим законом Рауля: підвищення температури кипіння і зниження температури замерзання розчину прямо пропорційні моляльній концентрації розчиненої речовини в розчині
де Сm — моляльна концентрація розчиненої речовини;
Е і К — відповідно ебуліоскопічна і кріоскопічна сталі розчинника.
Осмотичний тиск у розчині. Якщо розчини з різними концентраціями перегородити напівпроникною перетинкою, що має здатність пропускати тільки молекули розчинника і затримувати частинки розчиненої речовини, то почнеться одностороння дифузія (самочинне переміщення) молекул розчинника з розбавленого розчину до більш концентрованого (Рис.7).

Рис.7. Схематична ілюстрація явища осмосу
1-напівпроникна перетинка,
2-початковий рівень розчинів I і II (СII>CI),
3- рівноважний рівень розчинів
Явище проникнення молекул розчинника в розчин крізь напівпроникну перетинку називається осмосом. Щоб припинити осмос, потрібно ззовні до розчину прикласти тиск. Цей тиск викличе зворотний процес — вихід молекул розчинника з розчину. Тиск, який слід прикласти до розчину, щоб осмос припинився, називається осмотичним тиском.
За законом Вант-Гоффа: осмотичний тиск розчину (Росм.) прямо пропорційний молярній концентрації розчиненої речовини (См) та температурі (Т), отже:
Росм. = СмRT
Молярність См — це відношення числа молів розчиненої речовини n до об’єму V: 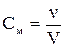 . Число молів розчиненої речовини n дорівнює масі цієї ре-човини m, поділеній на її молярну масу М. Підставивши значення См =
. Число молів розчиненої речовини n дорівнює масі цієї ре-човини m, поділеній на її молярну масу М. Підставивши значення См =  у рівняння Вант - Гоффа, знайдемо:
у рівняння Вант - Гоффа, знайдемо:
 |
Оскільки наведене рівняння за формою збігається з рівнянням стану ідеального газу (рівняння Менделєєва-Клапейрона), то закон Вант-Гоффа можна сформулювати так: осмотичний тиск розчину дорівнює такому газовому тиску, який мала б розчинена речовина в тому разі, коли була б переведена в газовий стан і займала такий самий об’єм, як і розчин.
У техніці використовують обернений осмос для очищення стічних вод, опріснення морської води тощо.
3.3. РОЗЧИНИ ЕЛЕКТРОЛІТІВ
Теорія електролітичної дисоціації. Ступінь електролітичної дисоціації. Електроліти — це речовини, які в розчинах та розплавах розпадаються на іони. Процес розпаду речовини на іони при розчиненні або при плавленні називається електролітичною дисоціацією. У розплавах дисоціюють лише сполуки з іонним типом зв’язку. Причиною руйнування кристалічних решіток та іонізації таких сполук є температура.
Теорія електролітичної дисоціації була розроблена шведським вченим С.Арреніусом (1859-1927), за що він у 1903р. одержав Нобелівську премію. Велика заслуга в розвитку теорії електролітичної дисоціації належить російським вченим Д.І.Менделєєву, І.О.Каблукову та українському вченому В.О.Кістяковскому. Згідно з нею при розчиненні електроліту у воді диполі води притягаються до полярних молекул або іонів розчиненої речовини. Полярні молекули (наприклад, НСl) в силовому полі поляризуються, зв’язок у них стає іонним, іони гідратуються і переходять у розчин (рис.8).

Рис.8. Схема електролітичної дисоціації полярної молекули
У розчинах встановлюється динамічна рівновага між числом молекул або іонних асоціатів, що розпалися на іони і знову утворилися, тобто дисоціація — процес оборотний, який записується в вигляді рівнянь електролітичної дисо-ціації:
HNO2<=>H+ + NO2-.
Іон водню H+ виявляє сильні акцепторні властивості й утворює з молекулою во-ди гідроксоній-іон:
H+ + Н2О ® Н3О+; DН = -711КДж.
Тому дисоціацію кислоти вірно було б записати з утворенням Н3О+:
HNO2 + Н2О<=>Н3О+ + NO2-.
Повнота перебігу процесу дисоціації характеризується ступенем дисо-ціації a, який визначається відношенням числа молекул електроліту, що розщепились у розчині на іони (nдис.), до загального числа молекул у розчині (n):
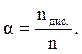
Ступінь дисоціації виражається в частках одиниці або в відсотках і змі-нюється від нуля до одиниці (0…100%). Величина ступеня дисоціації залежить від природи розчиненої речовини і розчинника, в якому розчинено електроліт, від температури та концентрації розчиненого електроліту.
За величиною ступеня дисоціації електроліти поділяють на сильні, слабкі та середньої сили.
Сильними є електроліти, ступінь дисоціації яких у 0,1н розчині переви-щує 30%. До сильних електролітів належать майже всі солі, деякі неорганічні кислоти (НСl, HBr, HJ, HNO3, H2SO4, HClO4 тощо), луги (LiOH, NaOH, KOH, RbOH, CsOH, Ca(OH)2, Sr(OH)2 ,Ba(OH)2 тощо).
Слабкими є електроліти, ступінь дисоціації яких у 0,1н розчині менший за 3%. До них належать вода, переважна більшість органічних і деякі неорганічні кислоти (H2CO3, H2SiO3, H2S, HCN тощо), нерозчинні гідроксиди металів та гідроксид амонію NH4OH.
Ступінь дисоціації електролітів середньої сили змінюється в інтервалі 3% — 30%. До них належать H2SO3, Н3РО4, HNO2, Мg(OH)2.
Найбільше значення ступеня дисоціації мають речовини з іонним зв’яз-ком. Для речовин з ковалентним полярним зв’язком a визначається ступенем полярності зв’язку: чим більша полярність, тим більше a. Речовини з кова-лентним неполярним зв’язком на іони не дисоціюють.
Дисоціація можлива лише в розчинниках з високим значенням діелект-ричної проникності:  ,
,  ,
,  тощо.
тощо.
Рівновага в розчинах слабких електролітів. У розчинах слабких електролітів та електролітів середньої сили одночасно присутні як молекули, так і іони, що перебувають у динамічній рівновазі, тобто процес дисоціації є оборотним. Тому процеси дисоціації молекул на іони описують за загальними законами рівноваги. Наприклад, для процесу
KmAn<=>mKn+ + nAm-
константу рівноваги або константу дисоціації можна описати рівнянням:
 |
Таким чином, константа дисоціації електроліту — це величина, що дорівнює відношенню добутку рівноважних концентрацій іонів, на які дисоціював елек-троліт, до рівноважної концентрації недисоційованих молекул електроліту з урахуванням стехіометричних коефіцієнтів.
Величина Кдис. не залежить від концентрації електроліту, а залежить тіль-ки від природи електроліту, природи розчинника та від температури. Тому константа дисоціації є більш загальною характеристикою, ніж ступінь дисоці-ації. Зрозуміло, що чим більша величина константи дисоціації, тим більш дисо-ційованою буде дана сполука.
 |
Константа дисоціації пов’язана зі ступенем дисоціації. Так, якщо загальну молярну концентрацію електроліту в розчині позначити С, то для бінарного
електроліту KA<=>K++A- рівноважні концентрації іонів К+ та А- будуть дорів-нювати aС, а рівноважна концентрація недисоційованих молекул — (С-aС), тоді
 |
Це рівняння є математичним виразом закону розведення Оствальда (1888р.) для електролітів середньої сили. Для слабких електролітів, де a ® 0 і (1-a) ® 1 закон набуває вигляду:
Це рівняння виражає залежність ступеня дисоціації слабкого електроліту від концентрації: зі збільшенням концентрації розчину електроліту ступінь його дисоціації зменшується, і, навпаки, розведення розчину призводить до підви-щення ступеня дисоціації, оскільки розведення розчину перешкоджає зворот-ному процесу — утворенню молекул з іонів. Введення однойменних іонів змі-щує рівновагу дисоціації слабких електролітів вліво, тобто a зменшується.
Константа дисоціації — термодинамічна величина. При сталих темпера-турі та тиску вона пов’язана зі зміною стандартної енергії Гіббса DG0 процесу дисоціації:
DG° = - RTlnKдис.
Дисоціація кислот, основ і солей у водних розчинах. Кислоти дисоціюють з утворенням катіонів Н+ та аніонів кислотного залишку. Процес дисоціації сильної одноосновної кислоти HNO3 можна описати рівнянням:
HNO3 ® Н+ + NO3-.
Наявністю іонів Н+ зумовлюються загальні властивості кислот.
Основи дисоціюють з утворенням катіонів металу і гідроксид-іонів ОН-:
KOH ® K+ + OH-.
Наявністю у розчинах основ гідроксид-іонів OH- зумовлюються загальні властивості основ.
Багатоосновні слабкі кислоти та багатокислотні слабкі основи дисоцію-ють ступінчасто, причому константа дисоціації за кожним ступенем завжди на кілька порядків нижча, ніж за попереднім. Наприклад:
H2CO3 <=>Н+ + НСО3ˉ К´дис=4,5·10-7
НСО3ˉ <=>Н+ + СО32ˉ К´´дис=4,8·10-11
Fe(OH)3<=>Fe(OH)2+ + OH-;
Fe(OH)2+<=>Fe(OH)2+ + OH-;
Fe(OH)2+<=>Fe3+ + OH-.
Сильні основи і кислоти дисоціюють за першим ступенем як сильні електроліти, а за другим – як електроліти середньої сили. Це слід відображати в запису рівнянь дисоціації:
Н2SO4→H+ + HSO4ˉ
HSO4ˉ <=> H+ + SO42ˉ
Солі — сильні електроліти, дисоціюють з утворенням катіонів основного залишку та аніонів кислотного залишку:
BaCl2 ® Ba2+ + 2Cl-.
Дисоціація кислих солей відбувається за схемою:
NaH2PO4 ® Na+ + H2PO4-;
H2PO4-<=>H+ + HPO42-;
HPO42-<=>H+ + PO43-.
Основні солі дисоціюють за схемою:
FeOHCl2 ® FeOH2+ + 2Cl-;
FeOH2+<=>Fe3+ + OH-.
Колігативні властивості розчинів електролітів. Крім здатності проводити струм, розчини електролітів мають і інші особливості, зокрема, у розчинах електролітів спостерігаються відхилення від законів Рауля і Вант-Гоффа. Наприклад, 0,1моляльний розчин NaCl замерзає не при –0,186°С, а при – 0,318°С. Для приведення у відповідність теоретичних і практичних результатів у формули, що пов’язують властивість розчину та його кон-центрацію, вводиться поправочний коефіцієнт і — ізотонічний коефіцієнт. Він показує, у скільки разів збільшилося число всіх частинок розчиненої речовини (Ср) внаслідок дисоціації порівняно з вихідним числом молекул (С):
 .
.
Ізотонічний коефіцієнт для розчинів електролітів має значення більше за одиницю. Він визначається для кожного розчину експериментальним шляхом, наприклад, за зниженням тиску пари або температури замерзання або за підвищенням температури кипіння розчину електроліту та осмотичним тиском відносно до тих же величин, обчислених без урахування дисоціації:
 .
.
Для встановлення зв’язку між ізотонічним коефіцієнтом та ступенем ди-соціації електроліту a, проаналізуємо концентрації іонів і молекул у розчині. Якщо молярна концентрація вихідного розчину — С, то при ступені дисоціації α і кількості іонів, що утворюються з однієї молекули електроліту, рівної n, рівноважна концентрація іонів у розчині становить Сіон = αСn. Тоді рівноважна концентрація недисоційованих молекул дорівнює Смол = С (1-α). Звідси концентрація всіх частинок у розчині Счаст. становить:
Счаст.= αСn + С(1-α).
Тоді  , або
, або
 звідки
звідки  .
.
Знайдене таким чином значення α для сильних електролітів виражає “уяв-ний” ступінь дисоціації. Тоді як за даними спектрального та рентгено-струк-турного аналізу у таких розчинах немає недисоційованих молекул, тобто α му-сить бути рівним 1.
Це протиріччя зумовлене міжіонними взаємодіями у розчинах сильних електролітів. Внаслідок високої концентрації іонів катіони і аніони зазнають взаємного притягнення, а іони одного знака заряду відштовхуються один від одного. Тому в розчині сильного електроліту кожний іон оточений так званою іонною атмосферою переважно з протилежно заряджених частинок. Це гальмує рух іонів і створює ефект неповної дисоціації електроліту. Саме тому замість концентрації іонів С використовують їхню активність аі, тобто умовну (ефек-тивну) концентрацію, відповідно до якої вони діють. Активність іона пов’язана з його молярною концентрацією в розчині Ci співвідношенням:
аі = fi ∙Ci,
де fі — коефіцієнт активності іона.
 Коефіцієнти активності іонів залежать від складу і концентрації розчину, від заряду та природи іонів, а також від температури. Залежність f від складу і концентрації розведеного розчину має вигляд:
Коефіцієнти активності іонів залежать від складу і концентрації розчину, від заряду та природи іонів, а також від температури. Залежність f від складу і концентрації розведеного розчину має вигляд:
lg f = - 0.5Z1∙ Z2  ,
,
де Z1, Z2 — заряди іонів;
I — іонна сила розчину.
Кількісно І розраховують за формулою:
І = 0,5 ∑Сi Zі2 ,
де Сi — концентрація і- того іону;
Zі — заряд і -того іону.
Рівновага в системі розчин — осад. Кількість іонів важкорозчинних сполук (деякі солі та гідроксиди) визначається не ступенем дисоціації, як для слабких електролітів, а розчинністю сполуки.
За законом діючих мас для насиченого розчину важкорозчинного силь-ного електроліту KmAn, який дисоціює у розчині за рівнянням:
KmAn(тв.)<=>KmAn (розч.) → mK n+ + nAm-,
константа рівноваги буде мати вигляд:
 .
.
Оскільки [KmAn] = const, то К[KmAn] = K',а значить і [Kn+]m·[Am-]n=K'.
Отже, добуток концентрацій (активностей) відповідних іонів з ураху-ванням коефіцієнтів у рівнянні реакції за сталих температури й тиску є сталою величиною. Цю величину називають добутком розчинності і позначають ДР. Він кількісно характеризує здатність електроліту розчинятися. Наприклад:
ДР СаСО3 =  9.3∙10-9; ДР СаSO4 = 6.1 ∙ 10-5; ДР AgI = 1.5 ∙ 10-16.
9.3∙10-9; ДР СаSO4 = 6.1 ∙ 10-5; ДР AgI = 1.5 ∙ 10-16.
З ДР випливає, що утворення осаду можливе, якщо добуток концентрацій іонів важкорозчинного електроліту перевищує його ДР, а розчинення осаду важко-розчинного електроліту відбувається, якщо добуток концентрацій іонів менший, ніж його ДР.
Реакції в розчинах електролітів. Згідно з теорією електролітичної дисоціації, реакції в розчинах електролітів відбуваються між іонами. Тому суть процесів у розчинах електролітів найбільш повно виявляється в іонно-молекулярних рівняннях. У таких рівняннях слабкі електроліти, малорозчинні та газоподібні речовини записуються в молекулярній формі, а сильні електроліти — у вигляді іонів, що входять до їхнього складу.
Якщо реакції в розчинах ідуть без зміни ступенів окиснення елементів (іонообмінні реакції), то вони відбуваються практично до кінця в таких ви-падках, коли утворюються малорозчинні або газоподібні речовини, а також слабкі електроліти. Це ствердження відоме як правило Бертоле. Наприклад:

 CaCl2 + Na2CO3 ® CaCO3 ¯ + 2NaCl;
CaCl2 + Na2CO3 ® CaCO3 ¯ + 2NaCl;
Ca 2+ + 2Cl- + 2Na+ + CO32- ® CaCO3 ¯ + 2Na+ + 2Cl-;
Ca 2+ + CO32- ® CaCO3¯.
Дисоціація води. Водневий показник. Вода — дуже слабкий електроліт, який частково дисоціює за схемою:
H2O<=>H+ + OH-.
Застосувавши до рівняння дисоціації води закон діючих мас, дістанемо:
 .
.
Оскільки на іони розкладається лише одна з приблизно 108 молекул води, то концентрацію молекул, що не розпалися, можна вважати рівною загальній концентрації молекул води. Концентрацію ж молекул H2O можна розрахувати, якщо поділити масу 1л води на її молярну масу:

Підставивши цю величину у вираз константи дисоціації води, матимемо:
[H+]∙[OH-] = 1,8·10-16 · 55,5 або
КН2О = [H+]∙[OH-] = 10-14,
де КН2О — іонний добуток води.
Оскільки згідно з рівнянням дисоціації води [H+] = [OH-], то через іонний добуток води можна визначити рівноважні концентрації іонів Н+ та ОН-.

[H+] = [OH-] = √10-14 = 10-7 (моль/л).
КН2О сильно залежить від температури і не залежить від концентрації іонів H+ і OH-. Це дозволяє розраховувати концентрацію будь-якого з них через кон-центрацію іншого в розчинах кислот і основ.
Якщо в нейтральному середовищі [H+] = [OH-] = 10-7 моль/л, то в кислому середовищі, де переважають іони H+, [H+] > [OH-], тобто [H+] > 10-7 моль/л. У лужному середовищі переважають іони ОН-, [H+] < [OH-], тобто [H+] < 10-7 моль/л.
Кислотність водних розчинів зручно виражати через десятковий лога-рифм молярної концентрації іонів Н+, взятий з протилежним знаком. Ця величина називається водневим показником, її позначають символом рН:
pH= - lg a H+, або рН = - lg [H+].
За аналогією з рН виведено гідроксидний показник — це десятковий логарифм молярної концентрації гідроксид-іонів, взятий з протилежним знаком:
pOH= - lg a OH-, або рOН = - lg [OH-].
Між рН та рОН існує зв’язок:
pH + pOH = 14.
Водневий показник визначає характер реакції середовища. Так, в нейт-ральному середовищі, де [H+] = 10-7 моль/л, pH = -lg 10-7 = 7.
В кислому середовищі рН < 7, у лужному рН > 7.
Регулюючи рН середовища можна добитися розчинення одних речовин і осадження інших. Наприклад, при пом’якшенні води гашеним вапном залеж-но від рН середовища можуть відбуватися процеси:
Mg(HCO3)2 + 2Ca(OH)2 ® Mg(OH)2¯ + 2CaCO3↓ + 2H2O (pH³10.3)
2Mg(HCO3)2 + 3Ca(OH)2 ® (MgOH)2CO3¯ + 3CaCO3↓+ 2H2O (pH£9.5).
Водневий показник визначають потенціометричним методом, який грун-тується на вимірюванні електродних потенціалів водневого або скляного елек-тродів, які перебувають у стані рівноваги з іонами Н+. Для цього використо-вуються прилади, які називаються рН-метрами.
Кислотно-основні індикатори. Одним з найпростіших методів визначення ак-тивності іонів Н+ є метод використання кислотно-основних індикаторів, які здатні змінювати колір залежно від рН розчину.
Індикаторами можуть бути слабкі органічні кислоти НInd та основи IndОН, молекули та іони яких мають різний колір. При введенні в досліджува-ний розчин індикатори дисоціюють за одним з наступних механізмів:
НInd<=>Н+ + Ind-;
IndОН<=> Ind+ + OН-.
Дата публикования: 2015-09-18; Прочитано: 11548 | Нарушение авторского права страницы | Мы поможем в написании вашей работы!
