 |
Главная Случайная страница Контакты | Мы поможем в написании вашей работы! | |
Основные стадии технол-кого процесса. Понятие лимитирующей стадии тех-кого процесса
|
|
Химико-технолог-й процесс представляет собой совокупность операций, позволяющих получить целевой продукт из исходного сырья. Все эти операции входят в состав трех основных стадий, характерных практически для каждого химико-технолог-го процесса.На первой стадии проводят операции, необходимые для подготовки исходных реагентов к проведению хим. реакции. Реагенты переводят в наиболее реакционноспособное состояние. Например, известно, что скорость хим-их реакций сильно зависит от тем-ры, поэтому часто реагенты до проведения реакции нагревают. Газообразное сырье для повышения эф-ти процесса и уменьшения размеров аппаратуры подвергают компримированию до опред-го давления. Чтобы устранить побочные явления и получить продукт высокого качества, исходное сырье подвергают очистке от посторонних примесей, пользуясь методами, основанными на различии физических свойств (растворимость в различных растворителях, плотность, тем-ры конденсации и кристаллизации и т.д.). При очистке сырья и реакционных смесей широко применяют явления тепло- и массообмена, гидромеханические процессы. Могут быть использованы и хим. методы очистки, основанные на хим-их реакциях, в результате которых ненужные примеси превращаются в легко отделимые вещ-ва.Подвод реагирующ-х компонентов в зону реакции совершается путем молекулярной диффузии или путем конвекции.Соответствующим образом подготовленные реагенты на следующей стадии подвергают хим-му взаимодействию, которое может состоять из нескольких этапов. В промежутках между этими этапами иногда необходимо вновь использовать тепло-массообменные и другие физические процессы. Например, при производстве серной кислоты диоксид серы частично окисляют до триоксида, затем реакционную смесь охлаждают, извлекают из нее путем абсорбции триоксид серы и вновь направляют ее на окисление. В результате хим-х реакций получают смесь продуктов (целевых, побочных, попутных) и непрореагировавших реагентов. Заключительные операции последней стадии связаны с разделением этой смеси, для чего вновь применяют гидромеханические, тепло- и массообменные процессы, например фильтрование, центрифугирование, ректификацию, абсорбцию, экстракцию и т.д. Продукты реакции направляют на склад готовой продукции или на дальнейшую переработку; непрореагировавшее сырье вновь используют в процессе, организуя его рецикл.На всех этапах, а особенно на заключительных, проводят также рекуперацию вторичных материальных и энергет-их ресурсов. Потоки газообразных и жидких вещ-в, попадающих в окруж. среду, подвергают очистке и обезвреживанию от опасных примесей. Твердые отходы либо направляют на дальнейшую переработку, либо размещают для хранения в безопасных для окруж. среды условиях.Лимитирующей стадией наз-ся самая медленная и идущая хуже всех.Чаще всего это первая стадия.Она определяет общую скорость химико-тенолог-го процесса.Таким образом, химико-технолог-ий процесс в целом — это сложная система, состоящая из единичных связанных между собой процессов (элементов) и взаимодействующая с окружающей средой.
4)Закон действующих масс. Равновесие технологич. процессов. Под воздействием подвода или отвода энергии в форме теплоты или работы происходит изменение состояния термодинамич. системы(значений термдинамич. парам-ров), называемыое термодинамич. процессом. Процесс представл-ий собой непрерывный ряд равновесных состояний наз-ют равновесными. При этом равновесным считают состояние к кот. приходит система при постоянных внешних условиях характериз-мое неизменностью во времени термодинамич. параметров и отсутствием в системе потоков вещества и теплоты. Устойчивое равновесие харак-ся общими условиями: 1.неизменностью равновесного состояния системы во времени при постоян. внешних условиях; 2.подвижностью равновесия(самопроизвольным восстановл-ем состояния равновесия после снятия внешнего воздействия вызвавшего отклонение системы от положения равновесия);3.динамич. хар-ом равновесия т.е. установлением и сохранением равновесия вследствие равенства скоростей прямого и обратного процессов;4.возможностью подхода к состоянию равновесия с двух противоположных сторон;5. мин-ым значением энергии Гиббса G в изобарно-изотермических и энергии Гельмгольца F в изохорно-изотермических процессах. Из этих общих условий выводится конкретные условия для хим. равновесия. В основе любого химико-технологического процесса лежит химич. превращение вещ-ва –хим. реакция. Теоретически все реакции обратимы, т. е. в зав-ти от условий могут протекать как в прямом, так и в обратном направлениях. Однако во многих из них равновесие смещено полностью в сторону продуктов реакции и обратная реакция практически не протекает. Поэтому технолог. процессы делят на обратимые и необратимые. Необратимые процессы протекают лишь в одном направлении.Все обратимые процессы стремятся к равновесию, при кот. скорости прямого и обратного процессов уравниваются, в результате чего соотношение компонентов во взаимодействующих системах остается неизменным до тех пор, пока не изменятся условия протекания процесса. При изменении таких технолог. параметров, как тем-ра, давление, концентрация реагирующих вещ-в, равновесие нарушается и процесс может протекать в том или ином направлении вновь до наступления равновесия.Количественно состояние равновесия описывается законом действующих масс (ЗДМ): при постоянной тем-ре и наличии равновесия отношение произведения действующих масс продуктов реакции к произведению действующих масс исходных веществ есть величина постоянная, называемая константой равновесия К. ЗДМ (закон равновесных концентраций) в математ. форме был вырожен в 1867г. Рассмотрим кинетич. вывод ЗДМ на примере гомогенной реакции aA+bB↔rR+sS. Скорость прямой реакции пропорц-на произведению концентраций реагентов A и B: w=k1cАа сBb, а скорость обратной реакции – произведению концентраций продуктов R и S: w=k2cRr сSs. Каждая концентрация возведина в степень равную стехиометрич-му коэф-ту компонента в хим. ур-и. Из условия равенства скоростей прямой и обратной реакции в момент хим. равновесия k1cА,eа сB,eb = k2cR,er сS,es получаем ЗДМ Kc= k1/k2= cА,eа сB,eb/ cR,er сS,es, где cА,e, сB,e, cR,e, сS,e – равновесные концентрации. Kc – константа равновесия; k1, k2 – константы скоростей. Константа равновесия не зависит от концентрации, т.к. изменение концентрации одного из участников реакции вызовет такие изменения концентр-ии всех остальных вещ-ств, что Kc сохранит свое числовое значение. Основное значение ЗДМ состоит в том, что он устанавл-ет связь между равновесными конц-ми всех реагирующих вещ-в. При анализе реакций, протек-х в газовой фазе константу равновесия выражают через порциональные давления: Kp= pА,eа pB,eb/ pR,er pS,es. Константу равновесия можно выразить также через отношения молярных долей N участников реакции: KN= NА,eа NB,eb/ NR,er NS,es. Константы равновесия Kp, Kc и KN связаны между собой соотношениями: Kp=Kc(R*T)∆n; Kp=KN*P∆n, где Т – абсолютная тем-ра; R – молярная газовая постоянная; P – общее давление газовой смеси; ∆n – изменение числа молей газообразных вещ-в участвующих в реакции. Для реальных систем константу равновесия выражают через летучести f или активности a.
5)Принцип Ле-Шателье. Влияние основных параметров технологического режима на равновесия. Положение равновесия всегда зависит от внеш. условий,а так как внеш.условия не могут сохранятся неизмен-ми,то равновесие рано или поздно нарушается(смещается).Влияние изменения внеш.условий на положение равновесия-принцип смещения равновесия, принцип Ле Шателье:если на систему,находящ-ся в устойчивом равновесии,воздействовать извне,изменяя какое-нибудь из условий,определяющих положение равновесия,то в системе усилиться то направление процесса,течение кот-го ослабляет влияние произведен-го воздействия,и положение равновесия сместиться в том же напрвлении.Рассм-м реакцию аА+bB=rR+sS,где реагенты A,B и продукты реакции R,S-идеаль.газы.При равновесии справедливо равенставо:ln(p rR,e p sS,e/ p aA,e p bB,e)=-∆G0/RT=ln KpЕсли под внешним воздействием изменится значение одного из членов равенства ∆G°/(RT) или (p rR,e p sS,e/ p aA,e p bB,e), оно нарушится и система выйдет из состояния равновесия. В результате система будет стремиться к достижению нового состояния равновесия с новыми значениями равновесных парциальных давлений реагентов и продуктов.Так как равновесие харак-ся равенством скоростей прямой и обратной реакций, можно сказать, что смещение равновесия происходит тогда, когда произведенное воздействие неодинаково влияет на скорости прямого и обратного процессов. Это нарушение равенства скоростей и приводит к переходу системы в новое состояние равновесия, при котором скорости прямой и обратной реакций опять станут равными, но будут отличаться от первоначальных значений. Влияние давления. Характер влияния давления на равновесие хим. реакций определяется знаком разности числа молей газообразных участников реакции ∆n или знаком изменения объема ∆V.Для газовых реакций, в которых число молей продуктов превышает число молей реагентов, т. е. ∆n >0, увеличение давления неблагоприятно. Смещению равновесия реакции вправо способствует снижение давления. Если же реакция протекает с уменьшением числа молей (∆n < 0), повышение давления целесообразно — оно смешает равновесие реакции в сторону образования продуктов. Чувствительность положения равновесия к изм-ям давления тем больше, чем большим изменением объема ∆V (или ∆n) сопровождается тот или иной процесс. Знач-ые изменения объема могут происходить только в реакциях, в которых участвуют газы, или в тех случаях, когда хотя бы один из компонентов находится в газообр. состоянии. Влияние инертного газа. Введение инертного газа в систему при р = const подобно эффекту уменьшения общего давления. Если реакция протекает с умен-ем числа молей (∆n < 0), разбавление инертным газом смещает равновесие реакции в сторону исходных реагентов. С увеличением числа молей (∆n > 0) равновесие смещается вправо. Поэтому в технолог. процессах, сопровождаемых хим. реакциями, для которых ∆n < 0, стремятся к уменьшению накопления инертных газов в системе.Выводы о влиянии инертного газа следуют и из закона Дальтона:pJ= NJp из которого видно, что эффект разбавления (уменьшения NJ) подобен эффекту снижения общего давления р в системе. Влияние концентрации. В соответствии с принципом Ле Шателье введение в равновесную систему дополнит-ых кол-в какого-либо вещ-ва вызывает смещение равновесия в том направлении, при кот. концентрация этого вещ-ва уменьшается. Поэтому введение избытка исходных вещ-в смещает равновесие вправо; введение избытка продукта вызовет смещение равновесия влево.Увеличивая концентрацию одного из реагентов (создавая его избыток), можно повысить степень превращения другого. Этим широко пользуются в хим. технологии, добиваясь полного превращения дорогостоящего компонента сырья. Влияние температуры. Направление смещения равновесия при изменении тем-ры зависит от знака теплового эффекта реакции. Повышение тем-ры всегда благоприятствует накоплению вещ-в,образующихся в данной реакции с поглощением теплоты, т. е. усиливает эндотермическое направление процесса. Понижение тем-ры действует в противополож. сторону, т. е. усиливает экзотермическое направление.При изменении тем-ры процесса равновесие смещается в направлении, для которого изменение энтропии имеет тот же знак, что и изменение тем-ры ∆Т. Учет знака теплового эффекта реакции (∆Н < 0) приводит к тому же выводу: повышение тем-ры смещает равновесие в сторону исходных реагентов (усиливает эндотермическое направление реакции), понижение тем-ры действует в противополож. направлении.
Следует отметить, что с изменением тем-ры равновесие смещается тем сильнее, чем большим тепловым эффектом сопровождается та или иная хим. реакция.Итак, применяя принцип Ле Шателье, можно, не выполняя термодинамич. расчеты, предсказать направление хим. реакций, т. е. качественно судить о состоянии их равновесия.
6)Основные хар-ки технологич.процессов.Степень превращения,избирательность,выход продукта.Матем. выражения для определения этих хар-к. Для оценки эф-ти отдельных этапов процесса необходимо помимо общих экономич. показателей использовать такие критерии эф-ти, которые более полно отражали бы хим. и физико-хим. сущность явлений, происходящих в отдельных ап-ах технолог-ой схемы.В качестве таких показателей принято прежде всего использовать степень превращения исходного реагента, выход продукта, селективность .. Степень превращения реагента показывает, насколько полно в химико-технолог. процессе используется исходное сырье- это доля исходного реагента, использованного на хим. реакцию.Степень превращения реагента J:Xj=nj,0 - nj,F / nj,0 = ∆nj / nj,0 где nj,0 — кол-во реагента J в исходной реакц. смеси; nj,F — кол-во реагента J в реакционной смеси, выходящей из ап-та или находящейся в реакторе; ∆nJ,— изменение кол-ва реагента J в ходе хим. реакции.Чаще всего в хим. реакции участвует не один, а два реагента (или даже больше). Степень превращения может быть рассчитана по первому, второму или третьему реагенту, причем в общем случае не обязательно получаются равные результаты.Если протекает реакция (аА+bB=rR+sS), то в соответствии с ее стехиометр. ур-ем изменения количеств ее участников ∆nJ, связаны между собой следующими соотношениями: ∆nА/а=∆nB/b=∆nR/r=∆ns/s.Степень превращ-я реагентов А и В: XА=nА,0 – nА / nА,0 = ∆nА / nА,0; Xв=nВ,0 – nВ / nВ,0 = ∆nВ / nВ,0 →XВ= (nА.0 / nВ,0)*XА / (а/b) Ур-е устанавливает связь между степенями превращения реагентов А и В и позволяет рассчитать неизвестную степень превращения одного реагента, зная степень превращения другого.Если nА.0 / nВ,0=а/b т. е. реагенты А и В взяты для проведения реакции в стехиометрич. соотношении, то степени превращения хА и хВ равны между собой: хА= хВ. Если nА.0 / nВ,0>а/b,т.е реагент А взят в избытке,то хА< хВ . Обычно при выборе первонач-го состава реакц. смеси берут в избытке более дешевый реагент (например, воздух, воду и т. д.) с целью повышения степени использования более ценного сырья.Не всегда возможно достичь полного использования реагента (т. е. условия х =1). Большинство хим. реакций обратимы. Для обратимых реакций при заданных условиях их осуществления предельным яв-ся состояние хим-го равновесия. Этому состоянию соответствует и предельно достижимая при данных условиях равновесная степень превращения: XА.е=nА,0 – nА.е / nА,0 = ∆nА.е / nА,0,где где nА.е — кол-во реагента А в условиях равновесия; ∆nА.е- изменение кол-ва реагента А к моменту наступления равновесия (максимально возможное при данных условиях осуществления хим-й реакции).Если первоначаль.кол-во продукта R равно нулю(nR,0 =0),то nR= ∆nА r/a= nА,0XАr/a. Выход продукта. Степень превращения харак-ет эф-ть проведения процесса с точки зрения использования исходного сырья, но этой величины не всегда достаточно для харак-ки процесса с точки зрения получения продукта реакции. Поэтому вводят еще один критерий эф-ти — Выход продукта -отношение реально полученного кол-ва продукта к максимально возможному его кол-ву, которое могло бы быть получено при данных условиях осущ-я хим-ой реакции.Выход продукта R через: ФR=nR/nR,max.Величина nR,max зависит от типа осущ-я хим-й реакции.Для простой необратимой реакции выход продукта и степень превращ-я реагента совпадают.Для обратимой хим-й реакции ФR= nR/nR,е= (nА,0XАr/a)/ (nА,0XА.еr/a)= XА/XА.е(для обратимых реакций выход продукта равен доле, которую составляет реально достигнутая степень превращения от равновесной для данных условий проведения реакции). Селективность(избирательность). Выход продукта хар-ет полученный результат как долю от предельно возможного. Целесообразно оценить и реальную ситуацию, т.е. дать колич-ую опенку зф-ти целевой реакции по сравнению с побочными взаимод-ми.Критерием для такой опенки яв-ся селек-ть. Ее тоже выражают в долях единицы или процентах.Полная, или интегральная, селективность φ— это отношение кол-ва исходного реагента, расходуемого на целевую реакцию, к общему кол-ву исходного реагента, пошедшего на все реакции (и целевую, и побочные): φ=∆nА,ЦЕЛ/∆nА,∑. Мгновенной, или дифферен-ой, селек-тью φ ' наз-ют отношение скорости превращения исходных реагентов в целевой продукт к суммарной скорости расходования исходных реагентов. Полная селек-ть по целевому продукту R может быть выражена через кол-во полученного продукта R и кол-во реагента А, суммарно израсходованного на реакцию.С учетом стехиометр. соотношений кол-во реагента А, вступ-го в реакцию образования целевого продукта, равно (a/r) nR.Тогда полная селек-ть: φ=[(a/r) nR]/ [nА,0- nА].Сущ-ет ур-е связи между выходом,селектив-тью и степенью превращ-я: ФR= φ(XА/XА.е).Отсюда следует,что при выборе условий проведения слож.хим. реакций недостаточно обеспечить только высокое значение степени превращ-я реагентов или только высокую селек-ть;высокое значение выхода целевого продукта опред-ся совокупностью этих критериев эф-ти.
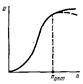
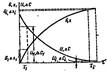 7)Изменение скорости реакции, степени превращения, движущей силы процесса во времени. Графическое выражение зависимости. Скорость технолог. процесса определяет производительность ап-ов и их кол-во в технолог-ой схеме. Скорость технолог. процесса обусловливается сочетанием скоростей прямой, обратной и побочных реакций, скоростью диффузии исходных вещ-в в зону реакции и продуктов хим. превращения из реакционной зоны, а также интенсивностью подвода (отвода) теплоты к взаимодействующим вещ-ам. Эффект-ть тепловых и диффузионных составляющих в основном определяется типом и конструкцией реактора, в котором осущ-ся данный технолог. процесс. Скорость технолог. процесса не может быть выше скорости хим-ой реакции, зависящей от природы реагирующих вещ-в, тем-ры, применяемых катализаторов и т. п., и лишь в пределе может стремиться к ней.Уровень приближения в основном и определяется конструкцией ап-та. В соответствии с законом действующих масс степень превращения (выход) х и общее кол-во продукта G, получаемого во времени изменяются по восходящей логарифмической кривой, представленной на рис. 13. Нисходящая кривая показывает изменение во времени скорости и и движущей силы процесса ∆ С. Кол-ва образующихся продуктов G1 и Gi, а соответственно и приращение выхода ∆ x1 и ∆ xi за равные промежутки времени τ1 и τi в начале и в конце процесса изменяются во много раз. Суммарная скорость процесса изменяется от и1 вначале до незначительной величины ut при приближении к равновесию.На практике нецелесообразно осущ-ть процесс с такой малой скоростью. Поэтому в обратимые процессы стараются проводить вдали от равновесия. Практически скорость процесса рассчитывается через константу скорости процесса k или на основе фактического выхода продукта Если кинетическое ур-е (основная формула скорости) процесса известно, то для количественной оценки интенсивности работы реакторов и для технолог-го расчета производ-ых процессов лучше пользоваться константой скорости процесса k, которая в гетерогенных процессах называется коэффициентом массопередачи. Коэф-т массопередачи измеряется обычно в килограммах вещ-ва, перешедшего из одной фазы в другую через 1 м2 поверхности раздела реагирующих фаз за 1 ч при разности действительной и равновесной концентраций,равной 0,1 МПа.Размерность k кг/(м2 * ч * Па) или кг*м3/(м2 * ч * кг) = м/ч. Константа скорости процесса не зависит от времени τ и концентрации реагирующих вещ-в С, а яв-ся лишь функцией тем-ры Т. Рис.13. Кинетика процесса соответствии с законом действующих масс(Р, Т=const): G – общее кол-во полученного продукта; x – выход продукта; u – скорость процесса; ∆ С – движущая сила процесса; τ – время.
7)Изменение скорости реакции, степени превращения, движущей силы процесса во времени. Графическое выражение зависимости. Скорость технолог. процесса определяет производительность ап-ов и их кол-во в технолог-ой схеме. Скорость технолог. процесса обусловливается сочетанием скоростей прямой, обратной и побочных реакций, скоростью диффузии исходных вещ-в в зону реакции и продуктов хим. превращения из реакционной зоны, а также интенсивностью подвода (отвода) теплоты к взаимодействующим вещ-ам. Эффект-ть тепловых и диффузионных составляющих в основном определяется типом и конструкцией реактора, в котором осущ-ся данный технолог. процесс. Скорость технолог. процесса не может быть выше скорости хим-ой реакции, зависящей от природы реагирующих вещ-в, тем-ры, применяемых катализаторов и т. п., и лишь в пределе может стремиться к ней.Уровень приближения в основном и определяется конструкцией ап-та. В соответствии с законом действующих масс степень превращения (выход) х и общее кол-во продукта G, получаемого во времени изменяются по восходящей логарифмической кривой, представленной на рис. 13. Нисходящая кривая показывает изменение во времени скорости и и движущей силы процесса ∆ С. Кол-ва образующихся продуктов G1 и Gi, а соответственно и приращение выхода ∆ x1 и ∆ xi за равные промежутки времени τ1 и τi в начале и в конце процесса изменяются во много раз. Суммарная скорость процесса изменяется от и1 вначале до незначительной величины ut при приближении к равновесию.На практике нецелесообразно осущ-ть процесс с такой малой скоростью. Поэтому в обратимые процессы стараются проводить вдали от равновесия. Практически скорость процесса рассчитывается через константу скорости процесса k или на основе фактического выхода продукта Если кинетическое ур-е (основная формула скорости) процесса известно, то для количественной оценки интенсивности работы реакторов и для технолог-го расчета производ-ых процессов лучше пользоваться константой скорости процесса k, которая в гетерогенных процессах называется коэффициентом массопередачи. Коэф-т массопередачи измеряется обычно в килограммах вещ-ва, перешедшего из одной фазы в другую через 1 м2 поверхности раздела реагирующих фаз за 1 ч при разности действительной и равновесной концентраций,равной 0,1 МПа.Размерность k кг/(м2 * ч * Па) или кг*м3/(м2 * ч * кг) = м/ч. Константа скорости процесса не зависит от времени τ и концентрации реагирующих вещ-в С, а яв-ся лишь функцией тем-ры Т. Рис.13. Кинетика процесса соответствии с законом действующих масс(Р, Т=const): G – общее кол-во полученного продукта; x – выход продукта; u – скорость процесса; ∆ С – движущая сила процесса; τ – время.
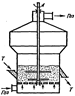
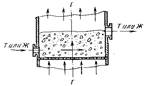 8)Характер изменения движущей силы процесса абсорбции газа жидкостью при прямоточной и противоточной подачи реактивов. Изменение концентрации компонентов различно в прямоточных, противоточных и перекрестных процессах; соответственно различны и формулы для вычисления движущей силы процесса. Применительно к газожидкостной гетерогенной системе направление движения реагирующих фаз в ап-те, например, с перекрестным током показано на рис.14. Перекрестный ток во взвешенном слое(в системе Г-Ж или Г-Т). Характер изменения движущей силы процесса при прямо- и противотоке изображен на рис.15. Движущая сила процесса ∆С = С-С* при прямотоке изменяется неравномерно по времени, которое определяется длиной пути совместного движения реагентов в аппарате. Для данного случая время пребывания определяется высотой насадки башни Н (время пропорционально высоте). Значение ∆С велико на входе в аппарат и стремится к нулю на выходе. Таким образом, и скорость процесса при прямотоке различна в различных точках ап-та. При противотоке движущая сила процесса в ходе его изменяется меньше. Если для прямотока при одном и том же значении ∆С в конце процесса Ск*< Сk, то при противотоке Ск*> Сk, а это значит, что при противотоке выход продукта значительно больше. Поэтому на практике всегда стремятся осуществить процесс по принципу противотока. Прямоток же применяют вынужденно.
8)Характер изменения движущей силы процесса абсорбции газа жидкостью при прямоточной и противоточной подачи реактивов. Изменение концентрации компонентов различно в прямоточных, противоточных и перекрестных процессах; соответственно различны и формулы для вычисления движущей силы процесса. Применительно к газожидкостной гетерогенной системе направление движения реагирующих фаз в ап-те, например, с перекрестным током показано на рис.14. Перекрестный ток во взвешенном слое(в системе Г-Ж или Г-Т). Характер изменения движущей силы процесса при прямо- и противотоке изображен на рис.15. Движущая сила процесса ∆С = С-С* при прямотоке изменяется неравномерно по времени, которое определяется длиной пути совместного движения реагентов в аппарате. Для данного случая время пребывания определяется высотой насадки башни Н (время пропорционально высоте). Значение ∆С велико на входе в аппарат и стремится к нулю на выходе. Таким образом, и скорость процесса при прямотоке различна в различных точках ап-та. При противотоке движущая сила процесса в ходе его изменяется меньше. Если для прямотока при одном и том же значении ∆С в конце процесса Ск*< Сk, то при противотоке Ск*> Сk, а это значит, что при противотоке выход продукта значительно больше. Поэтому на практике всегда стремятся осуществить процесс по принципу противотока. Прямоток же применяют вынужденно.
 Рис.15 Изменение концентрации при абсорбации газового компонента жидкостью при прямотоке (а) и противотоке (б): Сн, Сk – начальная и конечная концентрация абсорбируемого компонента в газе; Сk*, Сн* - начальная и конечная равновесные концентрации абсорбируемого компонента над его раствором.Так, при распылении мелко измельченного твердого материала или жидкости в потоке взаимодействующего с ними газа всегда получается прямоток, но при этом обеспечивается большая поверхность соприкосновения фаз. Примеры таких процессов встречаются при изучении производства серной кислоты, в крекинге нефтепродуктов и т. п. Сушку огнеопасных материалов также осуществляют в прямоточных сушильных аппаратах, так как при противотоке высушенный материал будет соприкасаться с горячими газами, что может вызвать воспламенение и даже взрыв.Как видно из рис. 15, концентрация реагирующих веществ при прямо- и противотоке изменяется в течение процесса по логарифмическим кривым. Соответственно средняя движущая сила процесса ∆Сср вычисляется как средняя логарифмическая из начальной ∆Сн и конечной ∆Сk движущих сил по формуле ∆Сср=∆Сн-∆Сk/2,3 lg(∆Сн/∆Сk). Для процесса абсорбации данная формула примет вид: а) при прямотоке ∆Сср= [(Сн- Сн*)-(Сk- Сk*)]/[2,3 lg((Сн- Сн*)/(Сk- Сk*))]; б) при противотоке ∆Сср= [(Сн- Сk*)-(Сk- Сн*)]/[2,3 lg((Сн- Сk*)/(Сk- Сн*))].
Рис.15 Изменение концентрации при абсорбации газового компонента жидкостью при прямотоке (а) и противотоке (б): Сн, Сk – начальная и конечная концентрация абсорбируемого компонента в газе; Сk*, Сн* - начальная и конечная равновесные концентрации абсорбируемого компонента над его раствором.Так, при распылении мелко измельченного твердого материала или жидкости в потоке взаимодействующего с ними газа всегда получается прямоток, но при этом обеспечивается большая поверхность соприкосновения фаз. Примеры таких процессов встречаются при изучении производства серной кислоты, в крекинге нефтепродуктов и т. п. Сушку огнеопасных материалов также осуществляют в прямоточных сушильных аппаратах, так как при противотоке высушенный материал будет соприкасаться с горячими газами, что может вызвать воспламенение и даже взрыв.Как видно из рис. 15, концентрация реагирующих веществ при прямо- и противотоке изменяется в течение процесса по логарифмическим кривым. Соответственно средняя движущая сила процесса ∆Сср вычисляется как средняя логарифмическая из начальной ∆Сн и конечной ∆Сk движущих сил по формуле ∆Сср=∆Сн-∆Сk/2,3 lg(∆Сн/∆Сk). Для процесса абсорбации данная формула примет вид: а) при прямотоке ∆Сср= [(Сн- Сн*)-(Сk- Сk*)]/[2,3 lg((Сн- Сн*)/(Сk- Сk*))]; б) при противотоке ∆Сср= [(Сн- Сk*)-(Сk- Сн*)]/[2,3 lg((Сн- Сk*)/(Сk- Сн*))].
9)Влияние тем-ры на скорость хим-х превращений.Правило Вант-Гофа,Ур-е Аррениуса. Влияние тем-ры на скорость химической реакции. Экспериментально при изучении кинетики хим. реакций было обнаружено, что при увеличении тем-ры на 10 градусов скорость реакции возрастает в 2—4 раза. Более строго эта зависимость выражается в виде уравнения Аррениуса:K=K0exp(-E/RT), где K— константа скорости реакции; K0— предэкспоненциальный множитель; Е— энергия активации реакции; R— универсальная газовая постоянная; Т— тем-ра. Энергия активации элементарной реакции Е— это минимальный избыток энергии над средней внутренней энергией молекул, необходимый для того, чтобы произошло хим. взаимодействие (энергетический барьер, который должны преодолеть молекулы при переходе из одного состояния реакционной системы в другое). Для обратимых реакции разность энергий активации прямой (E1) и обратной (E2) реакций равна тепловому эффекту реакции.
 Предэксп-ый множитель K0 учитывает число соударений, вероятность распада активированного комплекса реакции на исходные реагенты без образования продуктов реакции, пространственную ориентацию молекул реагентов, а также ряд других факторов, влияющих на скорость реакции и не зависящих от темп-ры. При более строгом рассмотрении следует учесть, что K0 также зависит от тем-ры, но при тем-ах, когда RT<<E, с достаточно хорошим приближением этой зависимостью можно пренебречь.Часто ур. Аррениуса представляют в виде линейной зависимости логарифма константы скорости от обратной тем-ры: ln K=f(1/T). В таком виде удобно провести его анализ.Из анализа зависимости ln K=f(1/T), приведенной на рис. 3.2, можно сделать следующие выводы.1) Из неравномерности температурной шкалы следует, что хим-ие реакции более чувствительны к изменениям тем-ры в области более низких температур. На прямой /, соответствующей хим-ой реакции с энергией активации 165 кДж/моль, выбраны два участка / и //, характеризующиеся одинаковым изменением тем-ры (∆T= 100 К), но в разных темпер-ых интервалах: участок / — в области тем-р, близких к комнатной, участок //— в области более высоких тем-р (600 К). Для участка / при изменении тем-ры на 100 градусов константа скорости реакции K1 увел-ся в 1,9 • 107 раз, для участка // при том же ∆Т наблюдается увеличение константы скорости лишь в 820 раз, примерно на четыре порядка ниже, чем на участке /.2)Второй важный вывод вытекает из сравнения тем-ных зависимостей скоростей реакций с различными значениями энергии активации. Чем выше энергия активации реакции, тем более чувствительна она к изменениям тем-ры.При изменении тем-ры от 500 до 600 К скорость первой реакции (E= 165 кДж/моль) увел-ся в 820 раз (участок //), а скорость второй реакции (E=40 кДж/моль) — лишь в 5,3 раза (участок ///). Последний вывод чрезвычайно важен при выборе условий проведения сложных (параллельных и послед-ых) реакций. Рис.3.2.Зависимость константы скорости хим. реакции от тем-ры для реакций с энергиями активации 165 кДж/моль (1) и 40 кДж/моль (2):участок I-изменение ln K при росте тем-ры с 300 до 400К;участк II-то же при росте тем-ры с 500 до 600К;участок III-изменение ln K для реакции с низкой энергией активации при изменении тем-ры с 500 до 600К.
Предэксп-ый множитель K0 учитывает число соударений, вероятность распада активированного комплекса реакции на исходные реагенты без образования продуктов реакции, пространственную ориентацию молекул реагентов, а также ряд других факторов, влияющих на скорость реакции и не зависящих от темп-ры. При более строгом рассмотрении следует учесть, что K0 также зависит от тем-ры, но при тем-ах, когда RT<<E, с достаточно хорошим приближением этой зависимостью можно пренебречь.Часто ур. Аррениуса представляют в виде линейной зависимости логарифма константы скорости от обратной тем-ры: ln K=f(1/T). В таком виде удобно провести его анализ.Из анализа зависимости ln K=f(1/T), приведенной на рис. 3.2, можно сделать следующие выводы.1) Из неравномерности температурной шкалы следует, что хим-ие реакции более чувствительны к изменениям тем-ры в области более низких температур. На прямой /, соответствующей хим-ой реакции с энергией активации 165 кДж/моль, выбраны два участка / и //, характеризующиеся одинаковым изменением тем-ры (∆T= 100 К), но в разных темпер-ых интервалах: участок / — в области тем-р, близких к комнатной, участок //— в области более высоких тем-р (600 К). Для участка / при изменении тем-ры на 100 градусов константа скорости реакции K1 увел-ся в 1,9 • 107 раз, для участка // при том же ∆Т наблюдается увеличение константы скорости лишь в 820 раз, примерно на четыре порядка ниже, чем на участке /.2)Второй важный вывод вытекает из сравнения тем-ных зависимостей скоростей реакций с различными значениями энергии активации. Чем выше энергия активации реакции, тем более чувствительна она к изменениям тем-ры.При изменении тем-ры от 500 до 600 К скорость первой реакции (E= 165 кДж/моль) увел-ся в 820 раз (участок //), а скорость второй реакции (E=40 кДж/моль) — лишь в 5,3 раза (участок ///). Последний вывод чрезвычайно важен при выборе условий проведения сложных (параллельных и послед-ых) реакций. Рис.3.2.Зависимость константы скорости хим. реакции от тем-ры для реакций с энергиями активации 165 кДж/моль (1) и 40 кДж/моль (2):участок I-изменение ln K при росте тем-ры с 300 до 400К;участк II-то же при росте тем-ры с 500 до 600К;участок III-изменение ln K для реакции с низкой энергией активации при изменении тем-ры с 500 до 600К.
10)Основные способы создания хороших условий контакта фаз в геторогентных процессах с участием твердой фазы. Основные способы создания хороших условий контакта фаз в гетерогенных процессах с участием твердой фазы можно разделить на четыре класса. 1)Перемешивание твердого материала на полках механич. мешалками. При этом материал размещается в ап-те на полках омывается сверху газом или жидкостью. Такими ап-ми в системе газ —твердое были обжиговые печи сернокислотного производства и цветной металлургии, а в системе твердое-жидкость-
ацетиленовый генератор. Для расчета поверхности соприкосновения реагирующих фаз в таких реакторах условно принимают общую площадь всех полок ап-та.2) Перемешивание твердого измельченного материала в потоке(в объеме) газа или жидкости. Ап-ты для создания таких условий перемешивания обычно представляют собой полые камеры или сосуды, в которые тяжелая фаза вдувается под давлением более легкой через специальные сопла или форсунки, и химическое превращение вещ-ва осущ-ся в турбулентном потоке газа или жидкости, который и обеспечивает интенсивное смешение фаз.Перемешивание в системе Ж-Т может осущ-ся также при помощи механич-их или пневмат-их мешалок. В этих случаях поверхность соприкосновения реагирующих компонентов достигает максимальной величины, равной общей (суммарной) поверхности всех твердых частиц, участвующих в превращении.3)Пропускание потока газа или жидкости через неподвижный слой кусков или гранул твердого материала, лежащего на колосниках или решетках. При этом происходит фильтрация газа или жидкости и потому такой слой называется фильтрующим. Ап-ты с фильтрующим слоем, как правило, просты по устройству, надежны в работе и широко распространены в промыш-ти. К основным типам ап-ов, работающих по принципу фильтрующего слоя, относятся колосниковые топки, шахтные и камерные печи, а также контактные аппараты. В реакторах фильтрующего слоя отсутствует интенсивное перемешивание. Поверхность взаимодействия фаз в фильтрующем слое меньше суммарной поверхности всех твердых частиц на величину, определяемую поверхностно соприкосновения соседних частиц друг с другом. Кроме того, в фильтрующем слое отсутствует перемешивание фаз и таким образом нет «обновления» поверхности твердого материала. Поэтому фильтрующий слой оказывается малоэффективным во многих гетерогенных процессах, особенно протекающих в диффузионной области. Для интенсификации диффузионных процессов стремятся применять другие виды перемешивания.
4. Перемешивание во взвешенном (кипящем, псевдоожиженном) слое. Взвешенный слой образуется при пропускании снизу вверх потока газа или жид-ти через слой твердого зернистого материала с такой скоростью, при которой частицы взвешиваются, начинают перемещаться, пульсировать или плавать в потоке более легкой фазы, как бы «кипеть». Поэтому в системах Г—Т такой слой наз-ют еще и кипящим. Скорость легкой фазы подбирается таким образом, чтобы частицы тяжелой фазы не покидали пределов реакционного ап-та. В этом случае, как и при перемешивании в объеме или в потоке, поверхность взаимодействия фаз яв-ся величиной, максимальной для данной степени измельчения материала. В то же время концентрация твердой фазы здесь выше, чем в ап-ах с распылением в объеме, поэтому в ап-ах со взвешенным слоем в системе газ — твердое достигается наивысшая интенсивность работы. Поверхность твердых частиц полностью омывается турбулентным потоком газа или жидкости. Скорость диффузионных процессов при этом резко возрастает. Поэтому взвешенный слой очень эффективен во многих гетерогенных технолог-их процессах.
11). Основные способы увеличения поверхности соприкосновения реагирующих фаз в системе газ – жидкость. Основные способы увеличения поверхности соприкосновения реагирующих фаз в системе Г-Ж также можно разделить на четыре класса.
1.Развитие поверхности жидкой фазы распределением жидкости в виде тонкой пленки по поверхности специальных приспособлений, называемых насадками. Ап-ты, в которых осущ-ся такой вид взаимодействия газов и жидкостей, наз-ся посадочными башнями или колоннами. Насадочные башни распространены в производствах серной, азотной, соляной и других кислот, некоторых солей, при химич. переработке топлива, в органическом синтезе и т. п. Для расчетов поверхность соприкосновения фаз принимается равной поверхности насадки, покрытой пленкой жидкости. Массопередача в таких ап-ах наз-ся пленочной, поскольку протекает на поверхности жидкой пленки. 2.Развитие поверхности жидкой фазы диспергированием или распылением, разбрызгиванием ее в полом ап-те пневматическим или механическим способом.Соответствующие ап-ты наз-ся башнями с разбрызг-ем или распылением жид-ти.Общая поверхность раздела фаз равна сумме поверх-ей всех капель жидкости. Массообмен здесь происходит на поверхности капли, поэтому такую массопередачу наз-ют капельной.3.Развитие поверхности взаимодействия реагирующих фаз за счет диспергирования газа в объеме жидкости. Этот метод увеличения поверхности наз-ся барботажем, так как газ барботирует (пробулькивает) в виде пузырей через слой жидкости в ап-ах с ситчатыми латками или колпачковыми тарелками. Иногда такие ап-ты наз-ют барботерами. Поверхность раздела фаз равна поверхности всех пузырьков и определяется периметром барботажа или площадью всех отверстий в решетках или тарелках. Массообмен в этих ап-ах происходит на поверхности газовых пузырей и поэтому массопередачу наз-ют пузырьковой. 4.Создание взвешенного слоя подвижной пены пропусканием газа снизу вверх через слой жидкости, так же как это наблюдается при псевдоожижении твердого материала. При этом также происходит взвешивание жидкости в потоке газа в виде пленок, струй и капель и интенсивное перемешивание их с пузырьками и струями газа. Газожидкостная система представляет собой слой подвижной (турбулентной) пены. Поверхность соприкосновения фаз характеризуется так называемой удельной поверхностью пены, которая яв-ся сложной функцией, зависящей от многих факторов и, как правило, определяемой экспериментальным путем.
12)Способы увеличения поверх-ти контакта в системах:а)две несмешивающ-ся жидкости;б)два твердых реагента. Процессы с участием двух и более твердых фаз (Т—Т) обычно представляют собой спекание твердых веществ при их обжиге.Спекание — это получение твердых пористых кусков из пылевидных или порошкообразных материалов при их нагреве до тем-ры ниже тем-ры плавления tпл. Реакции между твердыми вещ-ми без участия жидкой или газообразной фазы идут очень медленно из-за мало развитой межфазной поверхности F и малых скоростей диффузии. Фактически промышленные процессы спекания в смеси твердых веществ идут с участием газовой или жидкой фазы. Спекание применяется при агломерации руд в порошковой металлургии, в производстве глинозема (оксида алюминия); особенно широко процессы спекания используются в технологии силикатных материалов и изделий — вяжущих вещ-в, керамики, огнеупоров и др. Процессы, происходящие между реагентами в двух несмешивающихся жидких фазах (Ж—Ж), включают экстрагирование, эмульгирование и деэмульгирование. Экстрагирование основано на избирательной растворимости жидкостей в различных растворителях. Оно прим-ся в том случае, если ректификация жидкой смеси невозможна (низкая термич. стойкость, близость тем-р кипения компонентов и др.)- Экстрагирование исполь-ся при очистке нефтепродуктов, при извлечении фенола из надсмольных и сточных вод коксования и полукоксования, в производстве анилина, брома, иода. Эмульг-е — процесс диспергирования одной жидкости в другой, а деэмульгирование — расслоение эмульсий на исходные жидкости. Эмульсии и, следовательно, эмульгирование применяют в производстве лекарств, пищевых продуктов, пигментов и красок, а также для получения многих высокомолек-ых соед-й методом эмульсионной полимеризации. Примером деэмульгирования может служить обезвоживание нефти путем разрушения ее эмульсии с водой с применением ультразвука или других методов.Реакторы для гетерогенных жидкофазных процессов аналогичны гомогенным реакторам.Они обычно имеют перемешивающие устройства и внутренние теплообменники. В промыш-ти для взаимодействия несмешивающихся жидкостей исполь-ют реакторы периодич. и непрерывного действия, единичные и каскады. Экстрагирование осущ-ют также в колоннах с насадкой или ситчатыми полками, при противоточном режиме движения жидких фаз: тяжелая жидкость сверху вниз, а легкая — снизу вверх.Для лучшего контакта фаз также используют различ.мешалки.
13)Технологические системы с открытой цепью и замкнутые. Производство хим-их продуктов складывается из ряда хим-их и физических процессов, которые могут происходить последовательно или одновременно (параллельно) в одних и тех же ап-ах.Совокупность всех ап-ов, составляющих производство химич-го продукта, наз-ют химико-технолог-ой системой (ХТС). Взаимосвязь между ап-ами ХТС описывается математич-ой моделью, которая представляет собой систему ур-й, отражающих влияние технолог-го режима (концентраций, температур и других параметров режима) в предыдущих ап-ах на скорость процесса или режим работы в последующих. На основе математ-их моделей осуществляются автоматизированные системы управления производством (АСУ). Последователь. описание или изображение процессов и соответствующих им аппаратов, т. е. химико-технологической системой, наз-ся технолог.схемой производства.Бывают два типа: 1) с открытой цепью (разомкнутые); 2) циклические (замкнутые,круговые).Схемы с открытой цепью применяют в производствах, в которых по условиям равновесия основного химико-технолог-го процесса возможно достижение высоких степеней превращения (обычно x→1) без выделения продукта из реакционной смеси.Схема с открытой цепью состоит из ап-ов, через которые реагирующие компоненты проходят один раз.Если степень превращения в одном ап-те невелика, то включают в схему последовательно несколько однотипных ап-ов. Примером процесса с открытой цепью по газовой фазе может служить технолог-ая схема отделения (участка цеха) кислотной абсорбции нитрозных газов в производстве разбавленной азотной кислоты, которая приведена на рис. 19.Степень абсорбции оксидов азота в каждой башне относительно невелика, но в шести последовательных башнях суммарная степень извлечения оксидов азота из газов достигает приблизительно 92%.Оставшиеся нитрозные газы поглощаются щелочью в последующих башнях. Подобные технолог-ие схемы применяются в производствах серной и соляной кислот, некоторых минераль. солей и т. п.Химико-технологические системы, в которых равновесный выход в основном процессе невелик, осуще-ют по циклической схеме с выделением продукта после каждого прохода смеси через реакционную зону. Циклическая схема предусматривает многократное возвращение в один и тот же ап-т реагирующих масс или одной из фаз в гетерогенном процессе вплоть до достижения заданной степени превращения.Примером циклической схемы могут служить синтез аммиака,синтезы спиртов, моторного топлива и т. п. Эти синтезы характ-ся тем, что при осуществляемых в настоящее время основных параметрах технолог-го режима (тем-ры, давления, активность катализатора) по условиям равновесия экзотермич. реакций за один проход через реактор достигается малая степень превращения исходных вещ-в в целевой продукт.

| Остаточные газы |
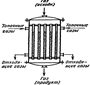 После выделения полученного продукта путем конденсации или абсорбции реакционная смесь вновь возвращается в реактор и циркулирует до превращения исходных компонентов близкого к полному.К циркулирующей смеси добавляется такое кол-во свежего сырья, которое равно расходу его на получение продукта за один цикл.В настоящее время циклич. схемы приобретают особое значение, поскольку,1-обеспечивают более высокую степень использования сырья, что улучшает экон-ие показатели процесса,2- в окр. среду выбрасывается меньшее кол-во вредностей.Во многих системах, основанных на гетерогенных процессах, применяют комбинированные технолог-ие схемы, в которых одна из реагирующих фаз последовательно проходит ап-ты, а другая совершает многократную циркуляцию через некоторые ап-ты системы.
После выделения полученного продукта путем конденсации или абсорбции реакционная смесь вновь возвращается в реактор и циркулирует до превращения исходных компонентов близкого к полному.К циркулирующей смеси добавляется такое кол-во свежего сырья, которое равно расходу его на получение продукта за один цикл.В настоящее время циклич. схемы приобретают особое значение, поскольку,1-обеспечивают более высокую степень использования сырья, что улучшает экон-ие показатели процесса,2- в окр. среду выбрасывается меньшее кол-во вредностей.Во многих системах, основанных на гетерогенных процессах, применяют комбинированные технолог-ие схемы, в которых одна из реагирующих фаз последовательно проходит ап-ты, а другая совершает многократную циркуляцию через некоторые ап-ты системы.
14)Основные положения, лежащие в основе расчетов материальных и тепловых балансов химико-технологических процессов. Чтобы харак-ть тот или иной технолог. процесс, составляют матер-ый и энергетический балансы.Составление матер. и энерг. балансов производят при проектировании новых производств и для анализа работы существующих. Матер. баланс — вещественное выражение закона сохранения массы вещ-ва,согласно которому во всякой замкнутой системе масса веществ, вступивших в реакцию, равна массе веществ, получившихся в результате реакции.Применительно к матер-му балансу любого технолог. процесса это означает, что масса веществ,поступивших на технолог-ую операцию-приход, равна массе всех веществ, получившихся в результате ее- расходу.Матер. баланс—зеркало технологического процесса.Матер. баланс составляют по ур-ю основной суммарной реакции с учетом параллельных и побочных реакций. Поскольку па практике приходится иметь дело не с чистыми веществами, а с сырьем сложного хим и механического состава,для составления матер. баланса приходится учитывать массу всех компонентов. Для этого пользуются данными анализов.Предположим, что технолог. процесс основан на хим. реакции, которая протекает по схеме mM+fF+dD →еЕ + lL+ ∆H,где М, F, D-исходное сырье; Е и L-основной и побочный продукты; m,d,f,е,I –стехиометрич. коэф-ты; ∆H- тепловой эффект хим. реакции. Матер. баланс чаще всего составляют на единицу массы готового (основного) продукта. Это значит, что для получения GE кг основного продукта необходимо израсходовать Gm, Gf и Gd кг сырья. При этом неизбежно получается CL кг побочного продукта. Тогда ур-е мат-го баланса будет GM+GF+GD= GE+GL+GП, где GП—непроизводительные затраты сырья и готового продукта, которые обусловлены неполнотой хим. превращений, а также механическими потерями при транспортировке и храпении сырья и готового продукта.Для расчета GE и GL применяют кинетические ур-я типа u= kv∆C или u =k∆C, u = kF∆C, u= dG/dτ=k’Pa+b.Все матер. расчеты сводятся в общую таблицу. Энергетический баланс составляют на основе закона сохранения энергии, в соответствии с которым в замкнутой системе сумма всех видов энергии постоянна. Обычно в химико-технолог. процессах составляется тепловой баланс.Применительно к тепловому балансу закон сохранения энергии может быть сформулирован: приход теплоты в данном цикле производства должен быть точно равен расходу ее в этом же цикле. При этом должна быть учтена вся теплота, подводимая в ап-т и выделяющаяся (поглощающаяся) в результате хим. реакции или физического превращения; теплота, вносимая каждым компонентом, как входящим в процесс или ап-т, так и выходящим из него, а также теплообмен с окр.средой.Тепловой баланс, как и матер., выражают в виде формул, таблиц и диаграмм. В ур-е теплового баланса входят следующие величины:1)QФ - физическая теплота материалов, поступающих в процесс ими ап-т, которая в некоторых справочниках наз-ся теплосодержанием материалов; 2) Qр - теплота, выделяющаяся в результате хим. реакции; 3)Qф.р - теплота, выделяющаяся в результате физических превращении вещества (физической абсорб-пии, адсорбции, кристаллизации и т. п.); 4)Qo - теплота, подводимая в ап-т (или процесс) извне, например обогрев. Перечисленные величины составляют статьи прихода.Расход теплоты составляют следующие величины:1)Q'ф - физическая теплота продуктов, выходящих из ап-та или процесса; 2) Q’p - теплота, которая затрачивается на эндотермическую реакцию;3) Q’ф.п -теплота, поглощающаяся в результате физических превращений (плавление, испарение, десорбция и т. п.); 4) Qп - потери теплоты в окр.среду. Таким образом, полное ур-е теплового баланса, составленное для одновременного протекания процессов с выделением (Qр, Qф.п) и поглощением (Q’p, Q’ф.п) теплоты, принимает вид Qф+Qp+ Qф.п+ Qo= Q’ф+ Q’p+ Q’ф.п+ Q’п. Теплоту, вносимую с материалом, обычно подсчитывают по ур-ю Qф=GсТ, где G-масса материала;с -средняя теплоемкость материала;Т- тем-ра.Подвод теплоты в ап-т (обогрев) Qo можно учитывать по потере теплоты теплоносителем, например для греющей воды Qо=Gc(Tн-Tк), для пара Qo=Gr, или по формуле теплопередачи через греющую поверхность Qо=ktF(Tг-Tх)*τ,, где Тн и Тк - начальная и конечная тем-ра воды; r - теплота парообразования; kT -общий коэф-т теплопередачи;F-поверхность, через которую осущ-ся теплопередача;Тг —тем-ра греющего вещ-ва; Тх—тем-ра нагреваемого (холодного) материала; τ —время теплопередачи. Потери теплоты в окр.среду рассчитывают по послед-му ур-ю.По данным матер-го и теплового балансов определяются также часовые потоки сырья, готовой продукции, побочных материалов и отходов производства; часовые расходы воды, теплоносителей, электроэнергии.
| Нитрозные газы |
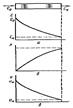 15)Классификация реактивов по режиму движения реагентов и по температурному режиму. Наиболее важными критериями для классификации хим. реакторов явл-ся: 1) непрерывность, или периодичность, операции; 2) режим движения и перемешивания реагентов; 3) температур. режим в реакционном объеме ап-та. Режим движения реагентов. Различают два предельных типа реакторов непрерывного действия: идеального вытеснения и полного (идеального) смещения (перемешивания). Реактор идеального вытеснения характ-ся тем, что реагенты последовательно «слой» за «слоем» без перемешивания ламинарным потоком проходят весь реакционный путь, определяемый, как правило, длиной (высотой) ап-та, которая обычно бывает значительно больше его диаметра. Время пребывания τ' любого элемента объема равно среднерасходному времени τср, определяемому по ур-ю τ'= τ ср=υ/Vp = H/ω, где v—реакционный объем ап-та;Vp—объемный расход реагентов; Н—высота (длина) реакционного пространства; ω —фиктивная, рассчитанная на полное сечение скорость потока. Интенсивность перемешивания реагентов обычно характеризуют диффузионным критерием Пекле: Peд = ωH/Dэ где Dэ —эффективный коэф-т диффузии, зависящий от коэф-в диффузии молекулярной — DM и турбулентной (конвективной) — DT. В реакторе идеального вытеснения перемешивание отсутствует, т. е. Dэ= 0 и диффузионный критерий Пекле Ре = ∞.
15)Классификация реактивов по режиму движения реагентов и по температурному режиму. Наиболее важными критериями для классификации хим. реакторов явл-ся: 1) непрерывность, или периодичность, операции; 2) режим движения и перемешивания реагентов; 3) температур. режим в реакционном объеме ап-та. Режим движения реагентов. Различают два предельных типа реакторов непрерывного действия: идеального вытеснения и полного (идеального) смещения (перемешивания). Реактор идеального вытеснения характ-ся тем, что реагенты последовательно «слой» за «слоем» без перемешивания ламинарным потоком проходят весь реакционный путь, определяемый, как правило, длиной (высотой) ап-та, которая обычно бывает значительно больше его диаметра. Время пребывания τ' любого элемента объема равно среднерасходному времени τср, определяемому по ур-ю τ'= τ ср=υ/Vp = H/ω, где v—реакционный объем ап-та;Vp—объемный расход реагентов; Н—высота (длина) реакционного пространства; ω —фиктивная, рассчитанная на полное сечение скорость потока. Интенсивность перемешивания реагентов обычно характеризуют диффузионным критерием Пекле: Peд = ωH/Dэ где Dэ —эффективный коэф-т диффузии, зависящий от коэф-в диффузии молекулярной — DM и турбулентной (конвективной) — DT. В реакторе идеального вытеснения перемешивание отсутствует, т. е. Dэ= 0 и диффузионный критерий Пекле Ре = ∞.
Рис.27. Реактор идеального вытеснения – контактный ап-т для эндотермических реакций с катализатором в трубках.
 Реактор полного смешения характ-ся тем, что частицы реагента (ион, молекула, или зерно твердого материала), попавшие в данный момент времени в ап-т, благодаря интенсивному перемешиванию имеет равную со всеми частицами вероятность первыми покинуть его.Здесь любой элемент объема мгновенно смешивается со всем содержимым реактора, так как скорость циркуляционных движений по высоте и сечению ап-та во много раз больше, чем скорость линейного перемещения по оси реактора.Эффективный коэф-т диффузии в этом случае Dэ = ∞ и в соответствии с уравнением Peд = ωH/Dэ критерий Ре = 0. Физической моделью реактора полного смешения может служить смеситель с пропеллерной или какой-либо иной интенсивной мешалкой.
Реактор полного смешения характ-ся тем, что частицы реагента (ион, молекула, или зерно твердого материала), попавшие в данный момент времени в ап-т, благодаря интенсивному перемешиванию имеет равную со всеми частицами вероятность первыми покинуть его.Здесь любой элемент объема мгновенно смешивается со всем содержимым реактора, так как скорость циркуляционных движений по высоте и сечению ап-та во много раз больше, чем скорость линейного перемещения по оси реактора.Эффективный коэф-т диффузии в этом случае Dэ = ∞ и в соответствии с уравнением Peд = ωH/Dэ критерий Ре = 0. Физической моделью реактора полного смешения может служить смеситель с пропеллерной или какой-либо иной интенсивной мешалкой.
Рис. 33. Реактор полного смешения апп-т с кипящим слоем зернистого материала с мешалкой.
Температурный режим. По тем-му режиму реакторы подразделяются на адиабатические, изотермические и политермические, называемые также программно-регулируемыми.
 1)Адиабатич. реакторы при спокойном (без перемешивания) течении потока реагентов не имеют теплообмена с окр. средой, т. е. снабжены хорошей тепловой изоляцией. Вся теплота реакции аккумулируется потоком реагирующих веществ. Скорость реакции на входе в ап-т (малые степени превращения) мала из-за низкой тем-ры системы, а на участках, близких к выводу, она также мала, так как степень превращения стремится к хр (или к единице). По типу, близкому к адиаб-му реактору вытеснения, работают контактные аппараты с фильтрующим слоем катализатора, камерные реакторы для осущ-я гомогенных превращений, прямоточные и др.2) Изотермические реакторы имеют постоянную тем-ру во всех точках реакционного объема, т. е. (tк = tср) во времени и пространстве. Скорость процесса опред-ся только концентрацией реагир. компонентов.Способы достижения изотермичности различны. Можно приблизиться к изотермическим условиям процесса при помощи теплообменных устройств, помещенных в реакц. объем (для отвода теплоты в экзотермических и подвода в эндотермических реакциях). При этом в каждом элементарном объеме ап-та отвод или подвод теплоты Qп должен быть равен теплоте реакции Qp, т. е. Qp=qpC0иxG=ktF∆tсрτ= Qп, где kt — коэф-т теплопередачи через теплообменную поверхность F при средней движущей силе ∆tcp за время τ. Изотермический режим достигается при интенсивном перемешивании реагентов а ап-те с мешалкой и в реакторах со взвешенным (кипящим, пенным) слоем, т. е. в ап-ах, в которых гидродинамический режим обеспечивает приближение к полному перемешиванию реагентов с продуктами реакции и инертными компонентами.Такие реакторы могут работать изотермически при регулировании тем-ры путем установки теплообменников (но без равенства (Qп и Qp) или же изотермически и адиабатически одновременно, когда во всем объеме тем-ра равна конечной. Изотермический режим приближенно соблюдается при малом значении λ, т. е. в реакторах с малой концентрацией исходных веществ и в реакциях с малым тепловым эффектом. В отдельных случаях изотермичность в реакторе достигается за счет теплового равновесия экзо- и эндотермического превращении экзотермической реакции. 3)Политермическими наз-ся реакторы, в которых теплота реакции лишь частично компенсируется за счет отвода (подвода) теплоты или процессов с тепловым эффектом, противоположным по знаку основному. Поскольку частичный подвод теплоты рассчитывается (программируется) при проектировании и может регулироваться при колебаниях режима, такие ап-ты называют также программно-регулируемыми. К политермическим ап-ам относят реакторы с малой степенью смешения реагирующих вещ-в и теплообменниками, помещенными внутри реакционного объема, например трубчатые контактные ап-ты.
1)Адиабатич. реакторы при спокойном (без перемешивания) течении потока реагентов не имеют теплообмена с окр. средой, т. е. снабжены хорошей тепловой изоляцией. Вся теплота реакции аккумулируется потоком реагирующих веществ. Скорость реакции на входе в ап-т (малые степени превращения) мала из-за низкой тем-ры системы, а на участках, близких к выводу, она также мала, так как степень превращения стремится к хр (или к единице). По типу, близкому к адиаб-му реактору вытеснения, работают контактные аппараты с фильтрующим слоем катализатора, камерные реакторы для осущ-я гомогенных превращений, прямоточные и др.2) Изотермические реакторы имеют постоянную тем-ру во всех точках реакционного объема, т. е. (tк = tср) во времени и пространстве. Скорость процесса опред-ся только концентрацией реагир. компонентов.Способы достижения изотермичности различны. Можно приблизиться к изотермическим условиям процесса при помощи теплообменных устройств, помещенных в реакц. объем (для отвода теплоты в экзотермических и подвода в эндотермических реакциях). При этом в каждом элементарном объеме ап-та отвод или подвод теплоты Qп должен быть равен теплоте реакции Qp, т. е. Qp=qpC0иxG=ktF∆tсрτ= Qп, где kt — коэф-т теплопередачи через теплообменную поверхность F при средней движущей силе ∆tcp за время τ. Изотермический режим достигается при интенсивном перемешивании реагентов а ап-те с мешалкой и в реакторах со взвешенным (кипящим, пенным) слоем, т. е. в ап-ах, в которых гидродинамический режим обеспечивает приближение к полному перемешиванию реагентов с продуктами реакции и инертными компонентами.Такие реакторы могут работать изотермически при регулировании тем-ры путем установки теплообменников (но без равенства (Qп и Qp) или же изотермически и адиабатически одновременно, когда во всем объеме тем-ра равна конечной. Изотермический режим приближенно соблюдается при малом значении λ, т. е. в реакторах с малой концентрацией исходных веществ и в реакциях с малым тепловым эффектом. В отдельных случаях изотермичность в реакторе достигается за счет теплового равновесия экзо- и эндотермического превращении экзотермической реакции. 3)Политермическими наз-ся реакторы, в которых теплота реакции лишь частично компенсируется за счет отвода (подвода) теплоты или процессов с тепловым эффектом, противоположным по знаку основному. Поскольку частичный подвод теплоты рассчитывается (программируется) при проектировании и может регулироваться при колебаниях режима, такие ап-ты называют также программно-регулируемыми. К политермическим ап-ам относят реакторы с малой степенью смешения реагирующих вещ-в и теплообменниками, помещенными внутри реакционного объема, например трубчатые контактные ап-ты.
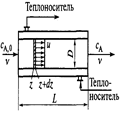 16)Изменение концентрации реагентов, степени превращения и скорости реакции в реакторе идеального вытеснения. Реактор идеального вытеснения представляет собой длинный канал, через который реакционная смесь движется в поршневом режиме(Рис 5.4.Схема реактора идеального вытеснения). Каждый элемент потока, условно выделенный двумя плоскостями, перпендик-ми оси канала, движется через него как твердый поршень, вытесняя предыдущие эл-ты потока и не перемешиваясь ни с предыдущими, ни со след-ми за ним эл-ми. Естественно, что при проведении хим. реакции, например реакции, в кот. учасвуют два или более реагентов перемешивание участников реакции явл. необход. услов-ем ее осуществл., иначе невозможным будет контакт между разноименными молек-ми, в результ. кот. и происходит элементарный акт реакции. Если в реакторе идеального перемешивания носит глоб-ый хар-р и благодаря ему парам. процесса полностью выравниваются по объему апп-та, в реакторе идеалного вытеснения перемеш-ие явл. локальным: оно происх. в каждом эл-те потока, а между соседними по оси реактора эл-ми перемеш-ия нет. Идеальное вытеснение возможно при выполн. допущений: 1) движущийся поток имеет плоский профиль линейных скоростей; 2) отсутств. перемеш-ие в направлении оси потока; 3) в каждом отдельно взятом сечении, перпенд-ом оси потока параметры процесса(концентр., температ., и т.д.) полностью выровнены. По длинне(высоте) изотермич. реактора монотонно уменьш. концентрац. реагентов и скорость реакции т.к. исходные реагенты расход-ся, а выход продукта увелич.(Рис 25 Изменение концентрации реагентов(а), степени превращения(б) и скорости реакции(в) в реакторе идеал. вытесн-ия).
16)Изменение концентрации реагентов, степени превращения и скорости реакции в реакторе идеального вытеснения. Реактор идеального вытеснения представляет собой длинный канал, через который реакционная смесь движется в поршневом режиме(Рис 5.4.Схема реактора идеального вытеснения). Каждый элемент потока, условно выделенный двумя плоскостями, перпендик-ми оси канала, движется через него как твердый поршень, вытесняя предыдущие эл-ты потока и не перемешиваясь ни с предыдущими, ни со след-ми за ним эл-ми. Естественно, что при проведении хим. реакции, например реакции, в кот. учасвуют два или более реагентов перемешивание участников реакции явл. необход. услов-ем ее осуществл., иначе невозможным будет контакт между разноименными молек-ми, в результ. кот. и происходит элементарный акт реакции. Если в реакторе идеального перемешивания носит глоб-ый хар-р и благодаря ему парам. процесса полностью выравниваются по объему апп-та, в реакторе идеалного вытеснения перемеш-ие явл. локальным: оно происх. в каждом эл-те потока, а между соседними по оси реактора эл-ми перемеш-ия нет. Идеальное вытеснение возможно при выполн. допущений: 1) движущийся поток имеет плоский профиль линейных скоростей; 2) отсутств. перемеш-ие в направлении оси потока; 3) в каждом отдельно взятом сечении, перпенд-ом оси потока параметры процесса(концентр., температ., и т.д.) полностью выровнены. По длинне(высоте) изотермич. реактора монотонно уменьш. концентрац. реагентов и скорость реакции т.к. исходные реагенты расход-ся, а выход продукта увелич.(Рис 25 Изменение концентрации реагентов(а), степени превращения(б) и скорости реакции(в) в реакторе идеал. вытесн-ия).
 17)Изменение концентрации реагентов, степени превращения и скорости реакции в реакторе полного смешения. Для модели идеал-го смешения принимается ряд допущений. Допуск., что в результ. интенсивного перемешив-ия устанавл-ся абсолютно одинак. усл-я в любой точке реактор: концентрации реагентов и продуктов, степени превращения реагентов, температ., скорость хим. реакции и т.д. Концентрация участников реакции в выходном потоке в момент времени τ строго равны концентрациям тех же вещ-ств в реакторе. Чтобы перечисленные допущения могли быть выполнены необход. принять еще одно: переход от одной концентрации к другой в реакторе идеального смешения не должен имеет протяженности во времени. Изменение концентрации исходного реагента от начальной во входном потоке в момент времени τ до концентрации в реакторе в этот же момент времени должно происходить мгновенно. Приблизиться к режиму идеального смешения можно обеспечив интенсивное перемешивание реакционной смеси мехонич-ми мешалками или циркуляционними насосами. Смешение близкое к идеальному легче выполнить в емкостных апп-ах с приблизит. равными диаметром и высотой. Изменение концентр. реагентов, степени превращения искорости превращ-я(u) по объему реактора полного перемешивания изображены на Рис. 30 Изменение концинтрац. (а), степени превращ-я (б) и скорости превращ-я(в) в реакторе полного перемештвания.
17)Изменение концентрации реагентов, степени превращения и скорости реакции в реакторе полного смешения. Для модели идеал-го смешения принимается ряд допущений. Допуск., что в результ. интенсивного перемешив-ия устанавл-ся абсолютно одинак. усл-я в любой точке реактор: концентрации реагентов и продуктов, степени превращения реагентов, температ., скорость хим. реакции и т.д. Концентрация участников реакции в выходном потоке в момент времени τ строго равны концентрациям тех же вещ-ств в реакторе. Чтобы перечисленные допущения могли быть выполнены необход. принять еще одно: переход от одной концентрации к другой в реакторе идеального смешения не должен имеет протяженности во времени. Изменение концентрации исходного реагента от начальной во входном потоке в момент времени τ до концентрации в реакторе в этот же момент времени должно происходить мгновенно. Приблизиться к режиму идеального смешения можно обеспечив интенсивное перемешивание реакционной смеси мехонич-ми мешалками или циркуляционними насосами. Смешение близкое к идеальному легче выполнить в емкостных апп-ах с приблизит. равными диаметром и высотой. Изменение концентр. реагентов, степени превращения искорости превращ-я(u) по объему реактора полного перемешивания изображены на Рис. 30 Изменение концинтрац. (а), степени превращ-я (б) и скорости превращ-я(в) в реакторе полного перемештвания.
Истинное время пребывания отдельной частицы в реакторе полного перемешивания может колебаться от 0 до ∞, а средн. время пребывания τср находится по ур-ю: τ'= τ ср=υ/Vp = H/ω, где v —реакционный объем аппарата; Vp—объемный расход реагентов; Н —высота (длина) реакционного пространства; ω —фиктивная, рассчитанная на полное сечение скорость потока. Скорость процесса можно представить как: u=G/τ=kcυ∆Ck, где υ – реакционный объем; ∆Ck – конечная движ-щая сила. Увеличение числа оборотов n смесительного устройства (или скорости потоков газа и жидкости) обеспечивает, с одной стороны, наибольшее приближение аппарата к реактору полного смешения, а с другой — к росту скорости процесса до некоторого предела (рис. 32 Зависимость скорости процесса u от числа оборотов n мешалки в реакторе полного смешения), при достижении, которого скорость превращения и может убывать. Оптимальное значение n соответствует такой степени превращения, при которой диффузионные сопротивления уже незначительны, но движущая сила процесса снижается вследствие усиления перемешивания. Поэтому чрезмерное увеличение скорости вращения мешалки может оказаться экономически нецелесообразным.
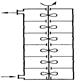
 18)Изменение концентрации основного исходного вещества по ступеням каскада реакторов полного смешения.
18)Изменение концентрации основного исходного вещества по ступеням каскада реакторов полного смешения.
Каскад представляет собой несколько последовательно соединенных проточных реакторов идеального смешения (Рис 5.9. Схема каскада реакторов идеального смешения)
Реакционная смесь проходит через все секции. Можно рассматривать в кач. примера такой модели не только систему послед-но располож. отдельных апп-ов, но и проточный реактор, разделенный внутри на секции. Например, близка к такому типу апп-та тарельчатая барботажная колонна (Рис. 5.10 Схема секционного апп-та с перемешиванием).
Для каскада реакторов идеального смешения должны выполняться допущения: 1) в каждой секции каскада выполн. условия реактора идеального смешения(мгновенное изменение парам-ов процесса, равенство парам-ов во всех точках секции и в потоке, выходящим из нее); 2) отсутствие обратного влияния: каждый последующий реактор не влияет на предыдущий. На рис. 5.11 сравнивается хар-тер изменения концентрации исходного реагента при прохождение реакционной смеси через различные реакторы. Рис 5.11. Изменение концентрации реагента при прохождении реакционной смеси через последовательные секции единичного реактора идеального смешения (1), реакторы идеального вытеснения (2) и каскада реакторв идеального смешения (3). В каскаде реакторов полного перемешивания состав реакционной смеси изменяется при переходе из одного апп-та в другой, а в каждом реакторе концентрационные и тем-ные поля безградиентны. Расчет каскада реакторов осущ-ся путем суммирования всех изменений, происход-щих в каждой ступени каскада. Поэтому концентрация основн. вещ-ва выходящего из N-го реактора каскада, для необротимой реакции первого порядка определяется как СИN=СИ0/(1+kCτ)NВыходной параметр для первой секции(концентрация) явл. входным параметром для второй секции.Расчет каскада реакторов идеаль. смешения обычно сводится к определению числа секций заданного объема необходимых для достиж. определенной глубины превращения, или к определению состава реакц/ смеси на выходе из i-ой секции каскада.
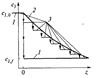
19)Общий вид и приемы поддержания темпер-го режима в адиабатич-х,изотермич-х и политермич-х ап-тах. По темпер-му режиму реакторы подразделяются на адиабатич., изотермич. и политермич.(программно-регулируемые). Адиабатич. реакторы при спокойном (без перемешивания) течении потока реагентов не имеют теплообмена с окр. средой, т. е. снабжены хорошей тепловой изоляцией.Вся теплота реакции аккумулируется потоком реагирующих вещ-в.Темпер-ый режим процесса в любой точке по высоте реактора опис-ся ур-ем:tK=tH± (∑qP *X/Gc) =tH±(CИ0qP/c),где tK, tH —конечная и начальная тем-ры системы; qp —тепловой эффект реакции (процесса) при полном превращении исходного вещ-ва или же при полном переходе основного компонента из одной фазы в другую в гетерогенных процессах; G — общее кол-во реакционной смеси, кг; с — средняя теплоемкость смеси в интервале тем-р tн — tк, кДж/(моль * К): Х— степень превращения.Это соотношение представляет собой ур-е прямой- tK= tH±λx, где λ – адиабатический коэф.процесса. Знак «+» соответствует протеканию экзо-, знак «-» —эндотермического процессов. Величину λ можно определить как тангенс угла наклона прямой (рис. 36).
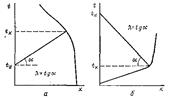 Рис. 36. Зав-ть конечной тем-ры процесса tк от степени превращения х для экзотермич. (а) и эндотермич. (б) реакций в адиабатическом реакторе идеаль. вытеснения.
Рис. 36. Зав-ть конечной тем-ры процесса tк от степени превращения х для экзотермич. (а) и эндотермич. (б) реакций в адиабатическом реакторе идеаль. вытеснения.

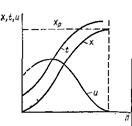 Рис. 37 Изменение тем-ры t, скорости реакции u и степени превращения х по высоте адиабатич. ректора идеально вытеснения. Скорость реакции на входе в ап-т (малые степени превращения) мала из-за низкой тем-ры системы, а на участках, близких к выводу, она также мала, так как степень превращения стремится к хр (или к единице).По типу, близкому к адиабатич-му реактору вытеснения, работают контактные ап-ты с фильтрующим слоем катализатора, камерные реакторы для осущ-я гомогенных превращений, прямоточные абсорберы с изолирующей футеровкой и др. Изотермические реакторы имеют постоянную тем-ру во всех точках реакционного объема, т. е. (tк = tср).Скорость процесса определяется только концентрацией реагирующих компонентов.Способы достижения изотермичности различны. Можно приблизиться при помощи теплообменных устройств, помещенных в реакционный объем (для отвода теплоты в экзотермич. и подвода в эндотермич. реакциях). При этом в каждом элементарном объеме ап-та отвод или подвод теплоты Qп должен быть равен теплоте реакции Qp, т. е. Qp=qpC0иxG=ktF∆tсрτ= Qп, где kt — коэф-т теплопередачи через теплообменную поверх-ть F при средней движущей силе ∆tcp за время τ. Изотермич. режим достигается при перемешивании реагентов а ап-те с мешалкой и в реакторах со взвешенным (кипящим, пенным) слоем, т. е. в ап-ах, в которых гидродинамический режим обеспечивает приближение к полному перемешиванию реагентов с продуктами реакции и инертными компонентами.Такие реакторы могут работать изотермически при регулировании тем-ры путем установки теплообменников (но без равенства (Qп и Qp) или же изотермически и адиабатически одновременно, когда во всем объеме тем-ра равна конечной. Изотермич. режим приближенно соблюдается в реакторах с малой концентрацией исходных вещ-в и в реакциях с малым тепловым эффектом. В отдельных случаях изотермичность в реакторе достигается за счет теплового равновесия
Рис. 37 Изменение тем-ры t, скорости реакции u и степени превращения х по высоте адиабатич. ректора идеально вытеснения. Скорость реакции на входе в ап-т (малые степени превращения) мала из-за низкой тем-ры системы, а на участках, близких к выводу, она также мала, так как степень превращения стремится к хр (или к единице).По типу, близкому к адиабатич-му реактору вытеснения, работают контактные ап-ты с фильтрующим слоем катализатора, камерные реакторы для осущ-я гомогенных превращений, прямоточные абсорберы с изолирующей футеровкой и др. Изотермические реакторы имеют постоянную тем-ру во всех точках реакционного объема, т. е. (tк = tср).Скорость процесса определяется только концентрацией реагирующих компонентов.Способы достижения изотермичности различны. Можно приблизиться при помощи теплообменных устройств, помещенных в реакционный объем (для отвода теплоты в экзотермич. и подвода в эндотермич. реакциях). При этом в каждом элементарном объеме ап-та отвод или подвод теплоты Qп должен быть равен теплоте реакции Qp, т. е. Qp=qpC0иxG=ktF∆tсрτ= Qп, где kt — коэф-т теплопередачи через теплообменную поверх-ть F при средней движущей силе ∆tcp за время τ. Изотермич. режим достигается при перемешивании реагентов а ап-те с мешалкой и в реакторах со взвешенным (кипящим, пенным) слоем, т. е. в ап-ах, в которых гидродинамический режим обеспечивает приближение к полному перемешиванию реагентов с продуктами реакции и инертными компонентами.Такие реакторы могут работать изотермически при регулировании тем-ры путем установки теплообменников (но без равенства (Qп и Qp) или же изотермически и адиабатически одновременно, когда во всем объеме тем-ра равна конечной. Изотермич. режим приближенно соблюдается в реакторах с малой концентрацией исходных вещ-в и в реакциях с малым тепловым эффектом. В отдельных случаях изотермичность в реакторе достигается за счет теплового равновесия
экзо- и эндотермического превращении, например компенсацией теплоты экзотермической реакции испарением растворителя (воды). Политермическими наз-ся реакторы, в которых теплота реакции лишь частично компенсируется за счет отвода (подвода) теплоты или процессов с тепловым эффектом, противоположным по знаку основному. Поскольку частичный подвод теплоты рассчитывается (программируется) при проектировании и может регулироваться при колебаниях режима, такие аппараты называют также программно-регулируемыми. К политермич. ап-ам относят реакторы с малой степенью смешения реагирующих веществ и теплообменниками, помещенными внутри реакционного объема, например трубчатые контактные аппараты. Тем-ра по высоте H таких ап-ов при осущ-ии экзотермических процессов изменяется по характерной кривой (рис. 38).
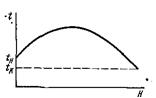 Рис.38. Изменение тем-ры по высоте политермич. реактора.
Рис.38. Изменение тем-ры по высоте политермич. реактора.
Дата публикования: 2015-10-09; Прочитано: 622 | Нарушение авторского права страницы | Мы поможем в написании вашей работы!
