 |
Главная Случайная страница Контакты | Мы поможем в написании вашей работы! | |
Лекция 5. Второе начало термодинамики
|
|
Второе начало термодинамики. Энтропия. Функция Гиббса. Изменение функции Гиббса при протекании химических реакций. Константа равновесия и функция Гиббса. Термодинамическая оценка вероятности протекания реакции.
Вторым началом термодинамики называется утверждение о том, что невозможно построение вечного двигателя второго рода. Закон получен опытным путём и имеет две эквивалентные друг другу формулировки:
а) невозможен процесс, единственным результатом которого является превращение всей теплоты, полученной от некоторого тела, в эквивалентную ей работу;
б) невозможен процесс, единственным результатом которого является передача энергии в форме теплоты от тела менее нагретого к телу более нагретому.
Функция δQ/T является полным дифференциалом некоторой функции S: dS=(δQ/T)обр(1) – эта функция S называется энтропией тела.
Здесь Q и S пропорциональны друг другу, то есть при увеличении (Q) (S) – увеличивается, и наоборот. Уравнение (1) соответствует равновесному (обратимому) процессу. Если процесс неравновесный, то энтропия увеличивается, тогда (1) преобразуется:
dS≥(δQ/T) (2) Таким образом, при протекании неравновесных процессов энтропия системы увеличивается. Если (2) подставить в первый закон термодинамики, то получим: dE≤TdS-δA. Его принято записывать в виде: dE≤TdS-δA’-pdV, отсюда: δA’≤-dE+TdS-pdV, здесь pdV – работа равновесного расширения, δA’- полезная работа. Интегрирование обеих частей этого неравенства для изохорно-изотермического процесса приводит к неравенству: A’V ≤ -∆E+T∆S (3). А интегрирование для изобарно-изотермического процесса (Т=const, p=const) – к неравенству:
A’P ≤ - ∆E+T∆S – p∆V=-∆H + T∆S (4)
Правые части (3 и 4) могут быть записаны как изменения некоторых функций, соответственно:
F=E-TS (5) и G=E-TS+pV; или G=H-TS (6)
F – энергия Гельмгольца, а G – энергия Гиббса, тогда (3 и 4) можно записать в виде A’V≤-∆F (7) и A’P ≤-∆G (8). Закон равенства соответствует равновесному процессу. При этом совершается максимально полезная работа, то есть (A’V)MAX=-∆F, и (A’P)MAX=-∆G. F и G называют соответственно изохорно-изотермический и изобарно-изотермический потенциалы.
Равновесие химических реакций характеризуется процессом (термодинамическим) при котором система проходит непрерывный ряд равновесных состояний. Каждое из таких состояний характеризуется неизменностью (во времени) термодинамических параметров и отсутствием в системе потоков вещества и теплоты. Равновесное состояние характеризуется динамическим характером равновесия, то есть равенством прямого и обратного процессов, минимальным значением энергии Гиббса и энергии Гельмгольца (то есть dG=0 и d2G>0; dF=0 и d2F>0). При динамическом равновесии скорость прямой и обратной реакций одинаковы. Должно также соблюдаться равенство:
µJdnJ=0, где µJ=(ðG/ðnJ)T,P,h=GJ – химический потенциал компонента J; nJ – количество компонента J (моль). Большое значение µJ указывает на большую реакционную способность частиц.
Скорости прямой и обратной реакций выражается:  =k1
=k1 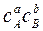
 и
и  =k2
=k2  , где с-концентрации; A, B – реагенты; R,S – продукты. В состоянии равновесия скорости равны и тогда получим выражение: kc =
, где с-концентрации; A, B – реагенты; R,S – продукты. В состоянии равновесия скорости равны и тогда получим выражение: kc =  =
= 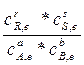 – константа равновесия, а выражение называется законом действующих масс (точнее концентраций).
– константа равновесия, а выражение называется законом действующих масс (точнее концентраций).
При анализе реакций, протекающей в газовой фазе концентрации можно заменить парциальными давлениями и выразить их через уравнение Менделеева-Клапейрона. В условиях химического равновесия в стандартном состоянии Тº=298 К и φº=0,098 МПа (1 атм):
∆Gº=-RTLn 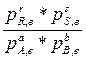 , обозначим выражение под знаком логарифма Кр (постоянная величина)-термодинамическая константа равновесия, получим:
, обозначим выражение под знаком логарифма Кр (постоянная величина)-термодинамическая константа равновесия, получим:
∆Gº=-RTLnКр (9)
Уравнение (9) называют уравнением изотермы Вант-Гаффа. Значение ∆Gº в таблицах в справочной литературе для многих тысяч химических соединений.
Можно рассчитать по формуле ∆Gº=∆Hº-Т∆Sº (10), решая совместно уравнения (9) и (10), получим:
Кр=e-∆Gº/(RT)= e-∆Hº/RT∙e∆Sº/R (11). Из (11) можно дать термодинамическую оценку вероятности протекания реакции. Так, для экзотермических реакций (∆Нº<0), протекающих с возрастанием энтропии, Кр>1, а ∆G<0, то есть реакция протекает самопроизвольно. Для экзотермических реакций (∆Нº>0) при убыли энтропии (∆Sº>0) самопроизвольное протекание процесса невозможно.
Если ∆Нº и ∆Sº имеют один и тот же знак, термодинамическая вероятность протекания процесса определяется конкретными значениями ∆Нº, ∆Sº и Тº.
Рассмотрим на примере реакции синтеза аммиака совместное влияние ∆Нo и ∆So на возможность осуществления процесса:
| N2 | + | 3H2 | ↔ | 2NH3 | |
| ∆Но298, кДж/моль | 2*(-46,1) | ||||
| Sо298,кДж/(моль*К) | 191,6 | 3*130,4 | 2*192,4 |
Для данной реакции ∆Нo298=-92,2 кДж/моль, ∆So298=-198 Дж/(моль*К), Т∆So298=-59кДж/моль, ∆Gо298=-33,2кДж/моль.
Из приведённых данных видно, что изменение энтропии отрицательно и не благоприятствует протеканию реакции, но в то же время процесс характеризуется большим отрицательным энтальпийным эффектом ∆Нº, благодаря которому и возможно осуществление процесса. С ростом температуры реакция, как показывают калориметрические данные, становится ещё более экзотермической (при Т=725К, ∆Н=-113кДж/моль), но при отрицательном значении ∆Sо повышение температуры весьма существенно уменьшает вероятность протекания процесса.
Дата публикования: 2015-09-18; Прочитано: 408 | Нарушение авторского права страницы | Мы поможем в написании вашей работы!
