 |
Главная Случайная страница Контакты | Мы поможем в написании вашей работы! | |
Превращения тирозина в надпочечниках и нервной ткани (синтез катехоламинов)
|
|
В мозговом веществе надпочечников и нервной ткани тирозин является предшественником катехоламинов (дофамина, норадреналина и адреналина) (см. схему Б на с. 509).
При образовании катехоламинов, которое происходит в нервной ткани и надпочечниках, и меланина в меланоцитах промежуточным продуктом служит диоксифенилаланин (ДОФА). Однако гидроксилирование тирозина в клетках различных типов катализируется различными ферментами:
· Тирозиназа в меланоцитах является Сu+-зависимым ферментом (см. выше).
· Тирозингидроксилаза (1) в надпочечниках и ка-техоламинергических нейронах не нуждается в ионах меди. Это - Fе2+-зависимый фермент, аналогично фенилаланингидроксилазе в качестве кофермента использующий Н4БП.
o Физиологическая роль тирозингидроксилазы чрезвычайно велика, так как этот фермент является регуляторным и определяет скорость синтеза катехоламинов.
o Активность тирозингидроксилазы значительно изменяется в результате:
o Аллостерической регуляции (ингибитор - норадреналин);
o Фосфорилирования/дефосфорилирования: в результате фосфорилирования с участием протеинкиназы А снижаются Кm для кофермента Н4БП и сродство фермента к норадреналину, в результате чего происходит активация тирозингидроксилазы.
o Количество фермента регулируется на уровне транскрипции.
· ДОФА-декарбоксилаза (2) (кофермент - ПФ) катализирует образование дофамина, который при участии дофамингидроксилазы (3) (монооксигеназы) превращается в норадреналин. Для функционирования фермента необходимы ионы Сu+, витамин С и тетрагидробиоптерин.
· В мозговом веществе надпочечников фенилэтаноламин-N-метилтрансфераза (4) катализирует метилирование норадреналина, в результате чего образуется адреналин. Источником метальной группы служит &АМ.
Дофамин и норадреналин служат медиаторами в синаптической передаче нервных импульсов,

Схема А
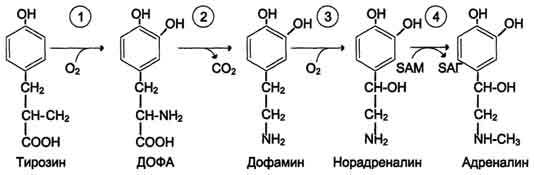
Схема Б
а адреналин - гормон широкого спектра действия, регулирующий энергетический обмен. Одна из функций катехоламинов - регуляция деятельности ССС (см. раздел 11).
3. Заболевания, связанные с нарушением
обмена фенилаланина и тирозина
Известно несколько наследственных заболеваний, связанных с дефектом ферментов обмена фенилаланина и тирозина в разных тканях.
Фенилкетонурия
В печени здоровых людей небольшая часть фенилаланина (∼10%) превращается в фенил-лактат и фенилацетилглутамин (рис. 9-30).
Этот путь катаболизма фенилаланина становится главным при нарушении основного пути - превращения в тирозин, катализируемого фенил-аланингидроксилазой. Такое нарушение сопровождается гиперфенилаланинемией и повышением в крови и моче содержания метаболитов

Рис. 9-30. Альтернативные пути катаболизма фенилаланина. При дефекте фенилаланингидроксилазы накопившийся фенилалан и н подвергается трансаминированию с а-кетоглутаратом. Образовавшийся фенилпируват превращается либо в фениллактат, либо в фенилацетилглутамин, которые накапливаются в крови и выделяются с мочой. Эти соединения токсичны для клеток мозга.
альтернативного пути: фенилпирувата, фенилацетата, фениллактата и фенилацетилглу-тамина. Дефект фенилаланингидроксилазы приводит к заболеванию фенилкетонурия (ФКУ). Выделяют 2 формы ФКУ:
· Классическая ФКУ - наследственное заболевание, связанное с мутациями в гене фенилаланингидроксилазы, которые приводят к снижению активности фермента или полной его инактивации. При этом концентрация фенилаланина повышается в крови в 20-30 раз (в норме - 1,0-2,0 мг/дл), в моче - в 100-300 раз по сравнению с нормой (30 мг/дл). Концентрация фенилпирувата и фениллактата в моче достигает 300-600 мг/дл при полном отсутствии в норме.
· Наиболее тяжёлые проявления ФКУ - нарушение умственного и физического развития, судорожный синдром, нарушение пигментации. При отсутствии лечения больные не доживают до 30 лет. Частота заболевания - 1:10 000 новорождённых. Заболевание наследуется по аутосомно-рецессивному типу.
· Тяжёлые проявления ФКУ связаны с токсическим действием на клетки мозга высоких концентраций фенилаланина, фенилпирувата, фениллактата. Большие концентрации фенилаланина ограничивают транспорт тирозина
и триптофана через гематоэнцефаличеекий барьер и тормозят синтез нейро-медиаторов (дофамина, норадреналина, серотонина).
· Вариантная ФКУ (коферментзависимая гиперфенилаланинемия) - следствие мутаций в генах, контролирующих метаболизм Н4БП. Клинические проявления - близкие, но не точно совпадающие с проявлениями классической ФКУ. Частота заболевания - 1-2 случая на 1 млн новорождённых.
· Н4БП необходим для реакций гидроксилирования не только фенилаланина, но также тирозина и триптофана, поэтому при недостатке этого кофермента нарушается метаболизм всех 3 аминокислот, в том числе и синтез ней-ромедиаторов. Заболевание характеризуется тяжёлыми неврологическими нарушениями и ранней смертью ("злокачественная" ФКУ).
Прогрессирующее нарушение умственного и физического развития у детей, больных ФКУ, можно предотвратить диетой с очень низким содержанием или полным исключением фенилаланина. Если такое лечение начато сразу после рождения ребёнка, то повреждение мозга предотвращается. Считается, что ограничения в питании могут быть ослаблены после 10-летнего возраста (окончание процессов миелиниза-ции мозга), однако в настоящее время многие педиатры склоняются в сторону "пожизненной диеты".
Для диагностики ФКУ используют качественные и количественные методы обнаружения патологических метаболитов в моче, определение концентрации фенилаланина в крови и моче. Дефектный ген, ответственный за фенилкетонурию, можно обнаружить у фенотипически нормальных гетерозиготных носителей с помощью теста толерантности к фенилаланину. Для этого обследуемому дают натощак ∼10 г фенилаланина в виде раствора, затем через часовые интервалы берут пробы крови, в которых определяют содержание тирозина. В норме концентрация тирозина в крови после фенилаланиновой нагрузки значительно выше, чем у гетерозиготных носителей гена фежилкетонурии. Этот тест используется в генетической консультации для определения риска рождения больного ребёнка. Разработана схема скрининга для выявления новорождённых детей с ФКУ. Чувствительность теста практически достигает 100%.
В настоящее время диагностику мутантного гена, ответственного за ФКУ, можно проводить с помощью методов ДНК-диагностики (рестрикционного анализа и ПЦР).
Тирозинемии
Некоторые нарушения катаболизма тирозина в печени приводят к тирозинемии и тирози-нурии. Различают 3 типа тирозинемии.
· Тирозинемия типа 1 (тирозиноз). Причиной заболевания является, вероятно, дефект фермента фумарилацетоацетатгидролазы, катализирующего расщепление фумарилацетоа-цетата на фумарат и ацетоацетат (рис. 9-28). Накапливающиеся метаболиты снижают активность некоторых ферментов и транспортных систем аминокислот. Патофизиология этого нарушения достаточно сложна. Острая форма тирозиноза характерна для новорождённых. Клинические проявления - диарея, рвота, задержка в развитии. Без лечения дети погибают в возрасте 6-8 мес из-за развивающейся недостаточности печени. Хроническая форма характеризуется сходными, но менее выраженными симптомами. Гибель наступает в возрасте 10 лет. Содержание тирозина в крови у больных в несколько раз превышает норму. Для лечения используют диету с пониженным содержанием тирозина и фенилаланина.
· Тирозинемия типа II (синдром Рихнера-Ханхорта). Причина - дефект фермента тирозина-минотрансферазы. Концентрация тирозина в крови больных повышена. Для заболевания характерны поражения глаз и кожи, умеренная умственная отсталость, нарушение координации движений.
· Тирозинемия новорождённых (кратковременная). Заболевание возникает в результате снижения активности фермента п-гидроксифенилпируватдиоксигеназы, превращающего п-гидроксифенилпируват в гомогентизиновую кислоту (рис. 9-28). В результате в крови больных повышается концентрация п-гидроксифенилацетата, тирозина и фенил-аланина. При лечении назначают бедную белком диету и витамин С.
Алкаптонурия ("чёрная моча")
Причина заболевания - дефект диоксигеназы гомогентизиновой кислоты (рис. 9-28). Дл
этой болезни характерно выделение с мочой большого количества гомогентизиновой кислоты, которая, окисляясь кислородом воздуха, образует тёмные пигменты алкаптоны. Это метаболическое нарушение было описано ещё в XVI веке, а само заболевание охарактеризовано в 1859 г. Клиническими проявлениями болезни, кроме потемнения мочи на воздухе, являются пигментация соединительной ткани (охроноз) и артрит. Частота - 2-5 случаев на 1 млн новорождённых. Заболевание наследуется по аутосомнорецессивному типу. Диагностических методов выявления гетерозиготных носителей дефектного гена к настоящему времени не найдено.
Альбинизм
Причина метаболического нарушения - врождённый дефект тирозиназы. Этот фермент катализирует превращение тирозина в ДОФА в меланоцитах. В результате дефекта тирозиназы нарушается синтез пигментов меланинов.
Клиническое проявление альбинизма (от лат. albus - белый) - отсутствие пигментации кожи и волос. У больных часто снижена острота зрения, возникает светобоязнь. Длительное пребывание таких больных под открытым солнцем приводит к раку кожи. Частота заболевания 1:20 000.
Нарушение синтеза катехоламинов (рис. 9-28) может вызывать различные нервно-психические заболевания, причём патологические отклонения наблюдаются как при снижении, так и при увеличении их количества.
Дата публикования: 2015-11-01; Прочитано: 1633 | Нарушение авторского права страницы | Мы поможем в написании вашей работы!
