 |
Главная Случайная страница Контакты | Мы поможем в написании вашей работы! | |
Механизмы гомогенного катализа
|
|
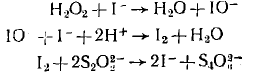
Каталитические реакции протекают по циклическому маршруту, т. е. по маршруту, состоящему из нескольких последовательных или последовательно-параллельных стадий, в результате которых один из компонентов — катализатор, расходуемый в первой стадии, вновь регенерируется в последней стадии. Остальные компоненты исходной реакционной смеси — субстраты каталитической реакции — в результате этого циклического маршрута превращаются в продукты реакции. Например, окисление тиосульфат иона перекисью водорода, которое катализируется нонами I, протекает по схеме:
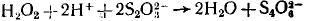
(вторая и третья стадии, по-видимому, не являются элементарными). Эта схема представляет собой циклический маршрут с итоговым уравнением
в двух первых стадиях которого катализатор расходуется, а в последней снова регенерируется.
В качестве второго примера можно привести реакцию хлорангидридов карбоновых
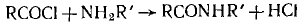
кислот с ароматическими аминами.
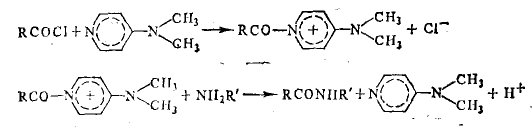
Она может быть существенно ускорена добавлением диметиламинопиридина. Это ускорение связано с возникновением циклического маршрута, приводящего к превращению субстратов в продукт реакции — амид — и к регенерации диметиламинопиридина
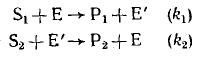
Общим в приведенных примерах является то, что в первой части маршрута один из субстратов реагирует с катализатором Е с образованием продукта превращения катализатора Е, а во второй части маршрута продукт превращения катализатора взаимодействует со вторым субстратом, превращая его в продукт реакции, с одновременной регенерацией катализатора.
Такой механизм катализа часто встречается в окислительно-восстановительных реакциях, и роль катализатора в этом случае сводится к созданию нового, более эффективного пути переноса электрона от восстановителя к окислителю.
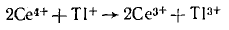
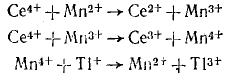
Наиболее отчетливо это видно на примере катализа реакций переноса электронов между ионами. Например, реакция идет очень медленно, так как требует одновременного участия трех ионов. Добавление ионов Мn2+ резко ускоряет процесс в результате возникновения маршрута, состоящего только из бимолекулярных Реакция с диметиламинпиридином представляет собой нуклеофильное замещение при карбонильном атоме С. В данном случае более сильный, чем ароматический амин, нуклеофил — диметиламинопиридин — обеспечивает быстрое превращение хлорангидрида в ацилдиметиламнно-лиридиний-катион, который благодаря наличию положительного заряда обладает высокой электрофильностью и легко атакуется амином. Такой тип катализа известен как нуклеофильный катализ.
Второй, наиболее распространенный механизм действия катализаторов включает в качестве первой стадии обратимое взаимодействие одного или нескольких субстратов с катализатором с образованием комплекса катализатор — субстрат. Так протекает катализ химических превращений ионами металлов и их координационными соединениями и катализ ферментами. К этому же типу можно отнести катализ кислотами, поскольку он включает, как правило, присоединение протона к одному из субстратов, что можно рассматривать как образование комплекса протон—субстрат.
Комплексообразование может приводить к нескольким различным эффектам, обеспечивающим ускорение реакции.
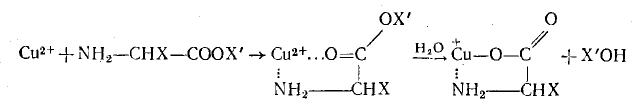
1. В комплексе с катализатором может происходить существенное перераспределение электронной плотности в молекуле субстрата, приводящее к изменению его реакционной способности. Например, присоединение к субстрату протона или образование субстратом координационной связи с ионом металла повышает электрофильность субстрата, делая возможным взаимодействие его с относительно слабыми нуклеофильными реагентами. Так, ионы Сu2+ являются эффективными катализаторами гидролиза эфиров аминокислот. Это прежде всего связано с тем, что последние образуют хелатный комплекс с ионом Сu2+, в котором положительный заряд иона Сu2+ поляризует связь С=О и облегчает нуклеофильную атаку молекулы воды на электрофильный атом углерода:
Поскольку ион металла при образовании координационной связи выступает как электрофильный компонент, этот тип катализа получил название электрофильного катализа.
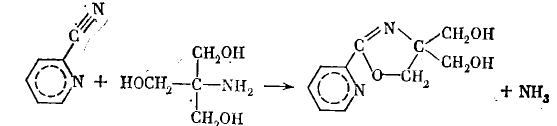
2. Если в образовании комплекса участвуют две молекулы субстрата, катализатор обеспечивает их пространственное сближение, благоприятное для протекания реакции. Например, ионы меди катализируют реакцию между нитрилом изоникотиновой кислоты и трис(оксиметил)-аминометаном (трисом), описываемую стехиометрическим уравнением.
Основными факторами при этом являются поляризация связи С=N в нитриле, облегчающая нуклеофильную атаку на атом (в результате этого ионы Сu+ являются катализаторами щелочного гидролиза нитрила), и одновременная координация обоих субстратов, обеспечивающая в лимитирующей стадии процесса атаку ОН-группы триса на поляризованный атом С нитрила (замыкание цикла с отщеплением аммиака происходит, по-видимому, в последующих стадиях, возможно, уже вне комплекса; стрелкой показано направление атаки атома О триса на атом С нитрила).
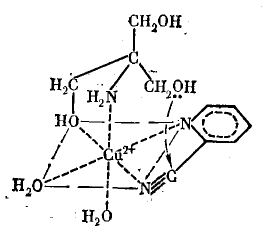
3. Помимо чисто пространственного эффекта сближения реагирующих групп, образование комплекса с катализатором может облегчить синхронное протекание разрыва и образования нескольких новых связей, необходимое для превращения молекул субстратов в молекулы продуктов. Например, это имеет место, когда для протекания реакции необходимо синхронное каталитическое участие и кислотной и основной групп. Так, превращение циклической формы 2,3,4,6-тетраметилглюкозы в открытую форму включает протонирование атома кислорода в цикле, расщепление связи С —О, синхронную передачу протона какому-либо основанию и образование двойной связи С=О. Обращение процесса может привести к изменению конфигурации при атоме С2 циклической формы (реакция мутаротации):
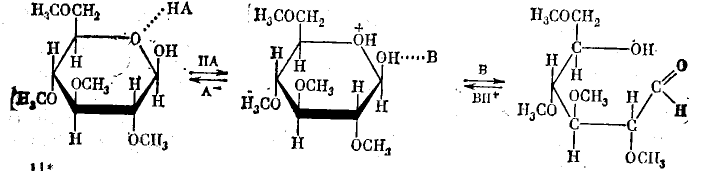
Реакция катализируется эквимолярной смесью фенола (кислота НА) и пиридина (основание В). Учитывая, что образовавшийся в комплексе катион пиридиния (ВH+) должен передать протон фенолят-иону (А-), легко видеть, что в этой реакции разрываются четыре связи и образуются четыре новые связи.
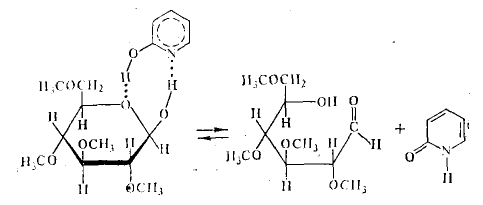
Гораздо более эффективным катализатором реакции мутаротации является α-оксипиридин, несмотря на то, что ОН-группа в этом соединении, выполняющая роль донора H+, менее кислая, чем ОН-группа фенола, а атом азота в α-оксипиридине, выполняющий роль акцептора протона, менее основен, чем в пиридине. Это случай бифункционального катализа. Протонирование атома кислорода циклической формы тетраметилглюкозы, разрыв связи С—О и отщепление протона от гидроксильной группы при атоме С с образованием двойной связи протекают синхронно в восьмицентровом циклическом активированном комплексе:
Наиболее полно и совершенно все перечисленные факторы, обеспечивающие воздействие катализатора на субстраты, используются в биологических катализаторах — ферментах. В настоящее время в результате успешного развития рентгеноструктурного анализа белков установлена полная пространственная структура ряда ферментов и их комплексов с субстратами. В качестве примера на рис. 1 приведена схема взаимодействия фермента карбоксипептидазы с субстратом.
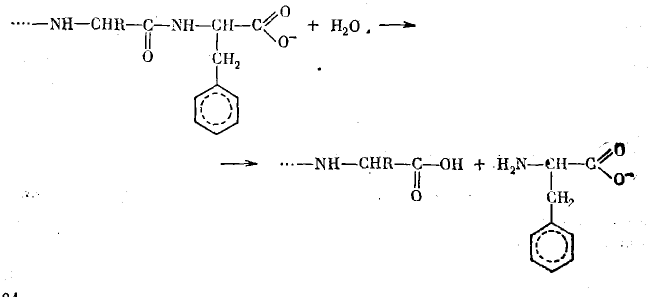
Карбэксипептидаза катализирует отщепление С-кониевой аминокислоты от пептидной цепи, причем наиболее эффективно отщепляются кислоты, содержащие гидрофобные ароматические остатки:
На рис. 1 изображен концевой фрагмент расщепляемой полипептидной цепи и функциональные группы фермента, принимающие то или иное участие в каталитическом процессе. Два имидазольных кольца (остатки аминокислоты гистидина) и карбоксильная группа остатка глутаминовой кислоты координированы с ионом цинка, заряд которого тем самым наполовину нейтрализован. Протонированная гуанидиновая группа (остаток аминокислоты аргинина) взаимодействует с ионизованной концевой карбоксильной группой субстрата. Этот же концевой аминокислотный остаток связан своим ароматическим кольцом с тремя гидрофобными радикалами фермента (остатки аминокислот изолейцина, тирозина и глутамина).
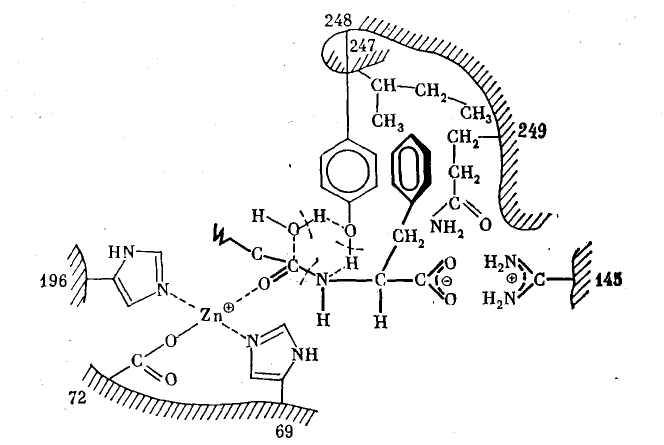
Рис. 1. Схема активного центра фермента карбоксипептидазы (по данным Липс-комба, Рика, Хартсака, Кешо и Бетджа):
Показаны фрагменты пептидном цепи с функциональными боковыми группами. Цифры обозначают порядковые номера остатков аминокислот, которым принадлежат эти функциональные группы. Молекула субстрата изображена с утолщенными связями. В шести-членном активированном комплексе штрихами показаны образующиеся связи, л сплошными линиями — разрывающиеся связи
В результате этих взаимодействий, которые закрепляют в двух точках С-концевой остаток субстрата, пептидная связь в случае, если С-концевая аминокислота представляет собой L-изомер, оказывается направленной на каталитический центр фермента, представленный ионом цинка и оксигруппой тирозина. Поляризация связи С=О ионом цинка облегчает нуклеофильную атаку молекулы воды на электрофильный атом С. Участие оксигруппы тирозина обеспечивает синхронное протекание разрыва трех связей и образования трех новых связей в циклическом шести центровом активированном комплексе.
На этом примере видны некоторые важнейшие черты, свойственные большому числу ферментов. Во-первых, катализатор имеет как бы два центра — связывающий (контактный) и собственно каталитический. Один из них, представленный в рассмотренном случае протонированной гуанидиновой группой и тремя гидрофобными радикалами, обеспечивает образование комплекса фермент — субстрат (связывание субстрата ферментом), в результате чего расщепляемая связь направляется на каталитический центр. Собственно каталитический центр представлен в рассмотренном случае ионом цинка и оксигруппой тирозина.
Во-вторых, на этом примере видны структурные основы высокой специфичности ферментов, в частности стереоспецифичности. Так, если бы С-концевая аминокислота была о-изомером, то в рассматриваемом случае в сторону каталитического центра оказался бы направленным атом Н, а не группа — NН—СО—, и каталитический процесс не смог бы произойти.
Из изложенного ясно также, почему фермент катализирует разрыв пептидной связи именно С-концевой аминокислоты и имеет преимущественное сродство к остаткам ароматических аминокислот. Действительно, именно взаимодействие заряда концевой карбоксильной группы и наличие гидрофобного ароматического остатка обеспечивает взаимодействие субстрата с контактным центром фермента, которое обеспечивает нужную ориентацию гидролизуемой связи относительно каталитического центра.
Дата публикования: 2015-10-09; Прочитано: 610 | Нарушение авторского права страницы | Мы поможем в написании вашей работы!
