 |
Главная Случайная страница Контакты | Мы поможем в написании вашей работы! | |
Приближенные методы расчета ΔG и константы равновесия
|
|
Для расчета термодинамических характеристик химических реакций по способам, изложенным в предыдущем разделе, нужно знать ряд величии, определяемых экспериментально; теплоемкость веществ и ее зависимость от температуры, тепловой эффект и т.п. В некоторых случаях одно или несколько необходимых исходных данных в литературе отсутствует; в этом случае приходится прибегать к менее точным методам расчета ΔG° и КР, не требующим, однако, постановки эксперимента, а основанным лишь на данных о структуре молекул реагирующих веществ и ее изменении в ходе реакции. Для ознакомления с методами расчета можно обратиться, например, к следующим монографиям:
1. Киреев В.А. Методы практических расчетов в термодинамике химических реакций. М., Химия, 1970.
2. Казанская А.С., Скобло В.А. Расчеты химических равновесий. М., Высшая школа, 1974.
В этих и других монографиях разбираются различные методы расчета термодинамических данных и наиболее применимые области их применения.
Равновесие в системах реальных газов
При выводе уравнения изотермы Вант-Гоффа используется уравнение состояния идеальных газов, а все изложенные выше методы расчета термодинамических потенциалов и констант равновесия пригодны лишь для газов, близких к идеальным, т. е. находящихся при низких давлениях и достаточно удаленных от состояния насыщенного пара. Между тем многие реакции органического синтеза часто проводятся при высоких давлениях, когда поведение реального газа существенно отклоняется от идеального. Для термодинамического расчета таких систем, чтобы сохранить общий вид термодинамических зависимостей, используют вместо парциальных давлений так называемые летучести, или фугитивности газов f, при постоянной температуре удовлетворяющие условию:
 (7.11)
(7.11)
При практических расчетах удобнее пользоваться активностью а, которая в общем случае представляет собой отношение летучести вещества в данном состоянии к его летучести f° в стандартном состоянии:
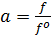 (7.12)
(7.12)
Для газов за стандартное принято состояние идеального газа при давлении
1 кгс/см2 ≈ 0,1 МПа. Поэтому для идеального газа активность равна его парциальному давлению, а для реального газа – его летучести. При повышенном давлении они могут существенно различаться, что учитывается коэффициентом активности γ, равным отношению летучести вещества к его парциальному давлению в газовой фазе:
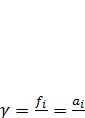 (7.13)
(7.13)
Поскольку при понижении давления все газы стремятся к идеальному состоянию, очевидно, что γ = 1 при Робщ → 0. Коэффициент активности однозначно связан с так называемым коэффициентом сжимаемости газа z, учитывающим его отклонение от уравнения состояния идеального газа: PV = zRT.
При постоянной температуре имеем:

Откуда
d(ln f) = z d(lnP)
d(ln f) – d(lnP) = ( z -1)d(lnP)
d(ln γ) = ( z -1)d(lnP)
Интегрирование последнего уравнения в пределах от 0 до Р дает:
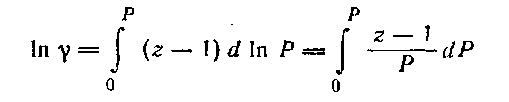
Это уравнение показывает, что коэффициент γ меняется с изменением давления и связан с коэффициентом сжимаемости z. Для идеальных газов z = 1 и, следовательно, ln γ = 0, т. е. γ = 1.
Принятое стандартное состояние в газовой фазе позволяет рассчитывать активность как произведение парциального давления на коэффициент активности, поскольку
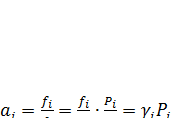 (7.14)
(7.14)
Уравнение изотермы Вант-Гоффа (7.4) в случае реальных газов преобразуется к виду:
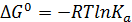 (7.15)
(7.15)
с учетом (7.14) можно написать:
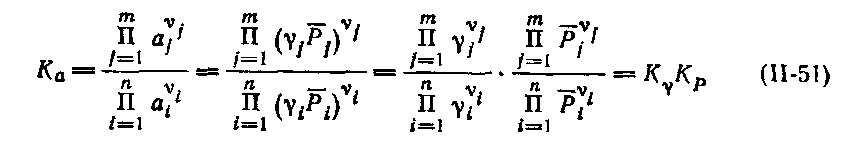
| (7.16) |
Здесь через Кγ обозначена первая дробь, представляющая собой по форме константу равновесия, в которой активности веществ заменены их коэффициентами активности. Уравнение (11-51) позволяет получить окончательное выражение изотермы Вант-Гоффа для реальных систем:
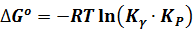 (7.17)
(7.17)
В нем Δ G° соответствует изменению стандартного изобарно-изотермического потенциала при Р = 1 кгс/см2 ≈ 0,1 МПа.
Таким образом, введение понятия активности позволяет свести учет неидеальности газов к вычислению некоторой величины Кγ, зависящей от стехиометрии реакции и коэффициентов активности реагентов. Последние находят из данных по сжимаемости реальных газов и выражают как функции приведенного давления π = Робщ/Ркр и приведенной температуры θ = Т/Ткр, где Ркр и Ткр – критические давление и температура данного газа (для водорода, гелия и неона в знаменателе должно стоять ( Ркр+8) и (Ткр+8)). Робщ – общее давление смеси, а не парциальное давление газа, так как его молекулы испытывают влияние и других компонентов смеси. Использование приведенных параметров, вытекающее из принципа соответственных состояний, позволяет применять один и тот же график или одинаковые табличные данные для расчета коэффициентов активности любых газов и паров. Табличные данные и графические зависимости приводятся в справочной литературе.
Экспериментальное определение равновесий
Когда точные данные по термодинамическим функциям или равновесию реакций отсутствуют, для их количественного расчета приходится прибегать к экспериментальному определению равновесий. Оно позволяет во многих случаях не только измерить константы равновесия, но и сделать полезные, а часто и наиболее точные расчеты других термодинамических функций – изменения изобарно-изотермических потенциалов, тепловых эффектов и т. д.
Методы экспериментального определения констант равновесия основаны на анализе состава реакционной смеси после достижения ею состояния химического равновесия. Для получения надежных результатов, кроме обычной проверки на воспроизводимость, необходимо ставить эксперименты таким образом, чтобы состояние равновесия достигалось с разных сторон, т. е. при первоначальном протекании как прямой, так и обратной реакции (в разных опытах). Только совпадение (в пределах ошибки опыта) аналитических данных по составу реакционной массы может в этом случае гарантировать, что изучается истинное равновесие.
Основные экспериментальные приемы для измерения констант равновесия делятся на две группы: 1) статические методы; 2) динамические методы.
При статических методах используют периодический реактор и ведут процесс до тех пор, пока результаты анализа реакционной массы не окажутся практически совпадающими. Начиная с этого момента, кинетические кривые для всех продуктов будут параллельны оси абсцисс, что и отвечает состоянию равновесия, исходя из которого, при известной стехиометрии, можно вычислить константу равновесия.
Динамические методы основаны на использовании непрерывных реакторов вытеснения и особенно удобны для изучения равновесия гетерогенных каталитических реакций. В этом случае постепенно увеличивают время пребывания реакционной смеси в аппарате, пока на выходе не будет достигнут практически постоянный состав, соответствующий равновесию. Основные трудности здесь такие:
- необходимость поддержания в реакторе строго заданной температуры, не меняющейся по длине (хотя бы на достаточно длинном участке перед выходом из реактора);
- необходимость резкого торможения реакции на выходе из аппарата – путем охлаждения или химического разложения, чтобы воспрепятствовать смещению равновесия при последующем хранении и анализе проб, так называемая – закалка.
Естественно, при динамических методах для более точного определения необходим ряд параллельных опытов с подсчетом средней константы равновесия и доверительного интервала ее величины.
Равновесие жидкофазных реакций
Расчет констант равновесия и составов равновесных смесей для гомогенных жидкофазных реакций или для взаимодействия газов с жидкостями представляет большой практический интерес, поскольку такие процессы часто встречаются в технологии органических соединений. Эти расчеты имеют ряд особенностей по сравнению с расчетами газофазных реакций, что обусловлено спецификой жидкого состояния веществ и растворов.
Приведенный нами ранее вид уравнения изотермы Вант-Гоффа (7.4) относится к газовой смеси. Однако можно показать, что такое же уравнение справедливо для любого состояния веществ:

| (7.18) |
Из уравнения (7.18) следует, что константа равновесия жидкофазной реакции, выраженная через активности, может быть рассчитана обычным путем – по изменению стандартного изобарно-изотермического потенциала. Для этого надо установить стандартное состояние для жидкостей и растворов и принять способ расчета или экспериментального определения активностей.
Для жидкостей стандартным принято их состояние в виде чистой жидкости при соответствующей температуре. Стандартные термодинамические параметры ряда жидкостей при 25 °С приведены в справочниках. Для пересчета на другие температуры можно воспользоваться методом Темкина – Шварцмана, считая, что теплоемкость постоянна (поскольку изменение температур для жидкофазных реакций невелико). Другой способ состоит в использований табличных или расчетных данных по стандартным изобарно-изотермическим потенциалам образования веществ для газообразного состояния Δ Gºf,Г. Поскольку они относятся к давлению газа (пара) 1 кгс/см2 (≈ 0,1 МПа), при их пересчете следует учесть Δ Gº пара при изменении давления от 1 кгс/см2 до давления, соответствующего равновесному давлению пара Р0 над жидкостью при данной температуре. Последующий перевод вещества из состояния пара в находящуюся с ним в равновесии жидкость не ведет к изменению Δ Gº. Таким образом, получаем:
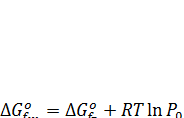 (7.19)
(7.19)
Эти данные оказываются вполне достаточными при термодинамическом расчете равновесий для идеальных систем, подчиняющихся законам Рауля и Генри. Поскольку активность равна отношению летучестей в данном и стандартном состоянии (a = f / f o), а летучесть вещества в растворе всегда равна летучести пара, находящегося с ним в равновесии (которую можно считать равной его парциальному давлению Pi), по закону Рауля имеем:
fж = Pi = Pi,0Ni
где Ni – мольная доля вещества в растворе. Отсюда получаем:

Таким образом, для идеальных растворов, когда стандартным принято состояние чистой жидкости, активность вещества равна его мольной доле, а константу равновесия следует выражать не через концентрации (как это часто принято), а через мольные доли:

| (7.20) |
В свою очередь, KN связана с Δ Gº известным соотношением:
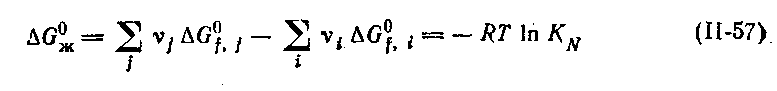
| (7.21) |
Основанные на изложенном методе расчеты равновесий жидкофазных реакций, дают вполне удовлетворительные результаты для смесей и растворов углеводородов и галогенпроизводных. В меньшей степени они справедливы для простых эфиров, сульфидов, меркаптанов, кетонов, альдегидов, аминов и нитросоединений – в реакциях их изомеризации, гидрирования, алкилирования и др., в которых другими реагентами или продуктами не являются вещества, способные нарушить идеальность системы.
Интересно выявить соотношения между константами равновесия в газовой и жидкой фазах для идеальных систем. Согласно предыдущему, имеем:
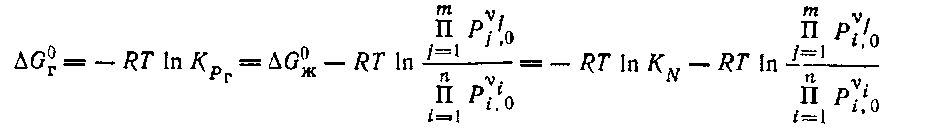
Откуда
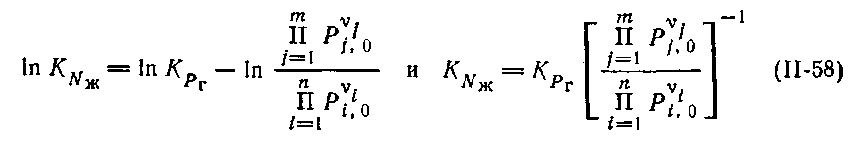
| (7.21) |
Следовательно, более простой путь для расчета равновесия в идеальных растворах состоит в определении изложенными ранее способами константы равновесия в газовой фазе (КP) и умножении ее на обратную величину отношения произведений равновесных давлений паров над чистыми жидкими продуктами и исходными реагентами, причем давления взяты в степенях, равных стехиометрическим коэффициентам. Так, например, для изомеризации н-бутана в изобутан стандартное изменение изобарно-изотермического потенциала равно Δ Gº298 = -900 кал/моль (-3767 Дж/моль), что дает константу равновесия для газовой фазы:
lgK P,298 = 900/(2,303·298·1,987) = 0,657
K P,298 = 4,56
Отсюда константа равновесия той же реакции в жидкой фазе равна:
 4,56
4,56  = 3,2
= 3,2
где 3,43 и 2,40 кгс/см2 – давления насыщенных паров изобутана и н-бутана при 25°С. Полученная величина КР близка к найденной экспериментально.
Для неидеальных растворов расчет равновесий существенно осложняется. Здесь стандартным остается для растворителя состояние чистой жидкости, а для растворенного вещества – гипотетическое состояние в растворе с активностью, равной единице, но с такими же термодинамическими и другими параметрами, которыми вещество обладает в бесконечно разбавленном растворе.
В реальных растворах активность растворенного вещества равна его мольной доле только при бесконечном разбавлении. Поэтому определение стандартных термодинамических параметров возможно, например, по экспериментальным данным о зависимости давления паров растворенного вещества над раствором при постепенном разбавлении. Эта зависимость подчиняется обобщенному уравнению
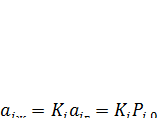 (7.22)
(7.22)
где активность пара  принята равной его парциальному давлению Pi,0. При бесконечном разбавлении записанная зависимость превращается в другую:
принята равной его парциальному давлению Pi,0. При бесконечном разбавлении записанная зависимость превращается в другую:
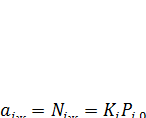 (7.23)
(7.23)
Откладывая на графике  против
против  (рис. 7.1) и экстраполируя кривую к
(рис. 7.1) и экстраполируя кривую к  , получают величину константы распределения Кi. После этого находят давление паров вещества над гипотетическим стандартным раствором с
, получают величину константы распределения Кi. После этого находят давление паров вещества над гипотетическим стандартным раствором с
аi = 1, т.е.  .
.

Изменение стандартного изобарно-изотермического потенциала образования растворенного вещества можно затем вычислить из ΔG0 f ,г с поправкой на изменение давления пара от  до 1 кгс/см2:
до 1 кгс/см2:
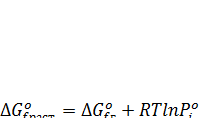 (7.24)
(7.24)
Например, для раствора ацетона в сероуглероде при 37 °С по зависимости давления паров ацетона от разбавления найдена Ki = 0,304 см2/кгс. Тогда давление его пара над стандартным раствором будет равно 1: 0,304 = 3,29 кгс/см2 и стандартный изобарно-изотермический потенциал раствора ацетона в сероуглероде составит:
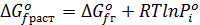 = -36 500 + RT ln 3,29 = -35 770 кал/моль =
= -36 500 + RT ln 3,29 = -35 770 кал/моль =
= -149 733 Дж/моль
Алгебраически складывая найденные значения ΔG0 f ,ж для всех компонентов, получаем изменение стандартного изобарно-изотермического потенциала реакции, известным образом связанное с Ка:
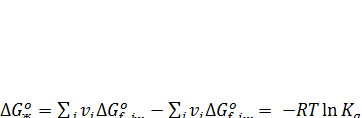
При таком расчете можно исключить из рассмотрения твердые компоненты, так как их активность всегда равна единице и Δ G0f = -RTlna = 0, а для реагента-растворителя нужно лишь ввести поправку, учитывающую, что его активность равна отношению давления паров над данным раствором и над чистой жидкостью:
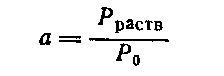
Для равновесий в системе газ – жидкость не требуется рассчитывать Δ G0f для растворенного газа, так как в этом случае пользуются другим выражением константы равновесия, включающим парциальные давления газообразных веществ.
Для неидеальных растворов, когда на термодинамические параметры компонентов влияет их взаимодействие не только с растворителем, но и друг с другом (образование комплексов, особенно за счет специфической сольватации между компонентами реакции), изложенные методы термодинамического расчета равновесий становятся непригодными. В этом случае требуется знать экспериментальные данные по изменению давления паров каждого вещества над многокомпонентными растворами при их постепенном разбавлении, что увеличивает объем необходимого исследования и усложняет расчеты.
В связи с изложенными трудностями термодинамического расчета жидкофазных реакций, для них бóльшее значение, чем для газофазных процессов, имеет экспериментальное определение равновесных составов смеси и констант равновесия. При этом непосредственно находят равновесные концентрации веществ, из которых легко рассчитать мольные доли и далее КС и KN. Эти результаты достаточны для определения состава равновесных смесей при любом соотношении исходных реагентов в случае идеальных систем. Однако принимать КС и KN постоянными для реальных растворов было бы ошибочным. Так, при кислотно-каталитической этерификации спиртов карбоновыми кислотами КС и KN существенно увеличиваются при повышении концентрации катализатора, что зависит от преимущественной сольватации протона более основной водой; это ведет к снижению действующей концентрации (активности) воды и смещению равновесия вправо. Точно так же при изомеризации ди- или полиалкилбензолов в присутствии хлористого алюминия с ростом концентрации AlCl3 увеличивается равновесное содержание 1,3- или 1,3,5-изомеров, образующих с катализатором более стабильные комплексы.
Эти примеры показывают, что для неидеальных систем экспериментально найденные мольные доли веществ при равновесии надо выразить через активности, чтобы определить величину действительно постоянной константы Кa.
При жидкофазных реакциях объем обычно меняется мало, и поэтому использованные ранее приемы расчета равновесных смесей в этом случае применимы для любых реакций, в том числе для идущих с изменением общего числа молей веществ. Как и раньше, равновесные концентрации выражают как функцию одной переменной – равновесной степени конверсии или равновесной концентрации ключевого вещества, которые находят затем из выражения для константы равновесия.
Вопросы и упражнения для самостоятельного решения
1. Какая разница между изменениями изобарно-изотермического потенциала ΔG и стандартного изобарно-изотермического потенциала ΔG°? Какой из них при равновесии равен нулю?
2. Чему равно изменение энтропии и изобарно-изотермического потенциала при равновесном превращении вещества из жидкости в пар при постоянных температуре и давлении? То же самое для химической реакции?
3. Объясните физический смысл, размерность и взаимные связи между летучестью, активностью, парциальным давлением и коэффициентом активности для реальных газов.
4. Каково соотношение между термодинамическими функциями одного и того же вещества, находящегося в стандартном состоянии в газе и растворе?
5. Для реакции А + Y = В имеем КP = 1,0. Какими двумя способами можно добиться, чтобы равновесная степень превращения вещества А достигла 90%?
6. Для последовательной реакции

выведите аналитическое решение для содержания (мольной доли) каждого вещества в равновесной смеси в зависимости от констант К1 и К2.
Дата публикования: 2015-07-22; Прочитано: 1450 | Нарушение авторского права страницы | Мы поможем в написании вашей работы!
